
Yuuki Hata
Assistant Professor, Department of Chemical Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology
He received his PhD Degree in polymer chemistry from Tokyo Institute of Technology in 2019 under the supervision of Professor Takeshi Serizawa. He was a postdoctoral researcher in the same group from April 2019 to March 2020 and then moved to the group of Professor Takamasa Sakai at the University of Tokyo. In August 2020, he became an Assistant Professor in the group of Professor Masayuki Ishihara and Professor Shingo Nakamura at National Defense Medical College, and then moved to the present post. His research interests include biomedical applications of self-assembled cellulose materials.

Takeshi Serizawa
Professor, Department of Chemical Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology
He received his PhD Degree in biotechnology from Tokyo Institute of Technology in 1996 under the supervision of Professor Toshinori Sato, who is now a Professor of Keio University. From 1996 to 2003, he worked as an Assistant and Associate Professor with Professor Mitsuru Akashi at Kagoshima University. He moved to the University of Tokyo in 2004 and managed his research group. Since 2011, he has been a full professor at Tokyo Institute of Technology. His current research focuses on the potential applications of bio(macro)molecules as functional materials.
Cellulose is a low-cost and versatile material and has long been used as paper and cloth. With the growing attention to the Sustainable Development Goals, recent studies have endeavored to expand the use of this representative biomass-derived polymer into highly functional materials. This article reviews our recent studies on the self-assembly of cello-oligosaccharides (i.e., short cellulose) for functionalizing conventional cellulose materials. Cello-oligosaccharides can be prepared by hydrolysis of naturally derived cellulose and by chemical synthesis via enzyme-catalyzed oligomerization. The former is a scalable method, and the resultant cello-oligosaccharides can be employed to add nanostructures to cellulose materials via self-assembly. The latter method enables us to synthesize cello-oligosaccharides with functional groups, the self-assembly of which allows for introducing functional groups as well as nanostructures into cellulose materials.
Cello-oligosaccharides can be prepared in a scalable manner by hydrolyzing naturally derived long-chain cellulose. Aqueous phosphoric acid solutions at concentrations of ~85% are known to act as a solvent for cellulose and to cause a phosphoric acid-catalyzed hydrolysis reaction. Thus, this solvent has long been used to prepare cello-oligosaccharides by dissolving cellulose in it and allowing the solutions to stand1–3. Recently, supercritical water has attracted attention for preparing cello-oligosaccharides4–6. Supercritical water hydrolyzes cellulose at an extremely high reaction rate; this reaction typically proceeds within several tens of milliseconds. Additionally, the preparation of cello-oligosaccharides using nonthermal atmospheric plasma combined with ball milling was reported7. In this method, ball milling reduced the crystallinity of cellulose and accelerated the depolymerization reaction by plasma.
Enzyme-catalyzed oligomerization has long been used to synthesize in vitro cello-oligosaccharides8–10. The representative enzymes used to cello-oligosaccharide synthesis are cellulase and cellodextrin phosphorylase (CDP). It is noted that the enzymes that synthesize cellulose in nature are rarely used for in vitro syntheses because of the low stability of the transmembrane proteins. Moreover, organic chemical synthesis of cello-oligosaccharides is rarely performed because synthesizing oligosaccharides requires advanced techniques for regioselective and stereoselective reactions of hydroxyl groups of saccharides.
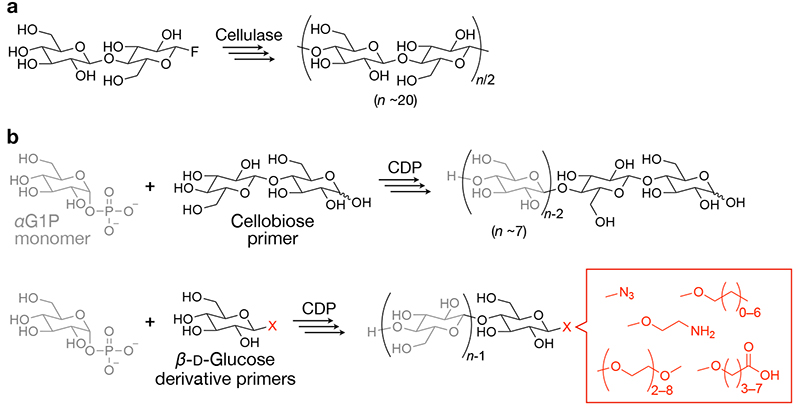
Cellulase-catalyzed oligomerization employs activated monomers, such as β-ᴅ-cellobiosyl fluoride11,12. Additionally, organic solvents, such as acetonitrile, were added to aqueous reaction solutions to deaccelerate the hydrolysis reactions catalyzed by cellulase, allowing condensation oligomerization with eliminating hydrogen fluoride (Figure 1a). The degree of polymerization (DP) of the resultant cello-oligosaccharides is typically ~20.
CDP phosphorolytically degrades water-soluble cello-oligosaccharides in nature13,14. A unique characteristic of the phosphorolysis reaction is its reversibility. The reversibility arises from the comparable free energies of the glycosidic linkage and the ester linkage in glycosyl phosphate13. Consequently, condensation oligomerization via the reverse phosphorolysis reaction proceeds in water by just tuning reaction conditions. For instance, relatively high concentrations (e.g., a few hundreds of millimolar) of α-ᴅ-glucose 1-phosphate monomer (glycosyl donor) facilitate oligomerization by CDP from cellobiose primer (glycosyl acceptor) (Figure 1b, upper panel)15,16.
Unique characteristics of CDP-catalyzed oligomerization include that various β-ᴅ-glucose derivatives as well as cellobiose can serve as primers. The use of β-ᴅ-glucose derivatives as primers produces cello-oligosaccharides having functional groups at the terminal (Figure 1b, lower panel). Our and other groups have investigated the recognition of β-ᴅ-glucose derivatives as primers by CDP and demonstrated the synthesis of various cello-oligosaccharide derivatives having azide17, alkyl18, amino19, carboxy20, and oligoethylene glycol groups21,22.
Cello-oligosaccharides with DPs ≥7 are almost water insoluble15,23,24; they self-assemble from a dissolved state in water and form nanostructures. During the CDP-catalyzed oligomerization reactions described above, growing cello-oligosaccharide chains self-assemble in situ when they have become water insoluble15,25. On the other hand, the use of appropriate solvents enables us to conduct the self-assembly of pre-prepared cello-oligosaccharides. Our group has focused on the dissolution in aqueous sodium hydroxide solution and subsequent neutralization as a fully aqueous system for cello-oligosaccharide assembly26–28. Cello-oligosaccharides have hydroxy groups with pKa values of ~13.5 and thus dissolve in an aqueous alkaline solution at pH ~14 by ionization through deprotonation29–31. Subsequent neutralization by adding an acid triggers the self-assembly of cello-oligosaccharides due to their reprotonation to be neutral and water-insoluble. We call this system "neutralization-induced self-assembly". Aqueous phosphoric acid solutions can also be used as solvent for cello-oligosaccharide assembly with using water as coagulant32.
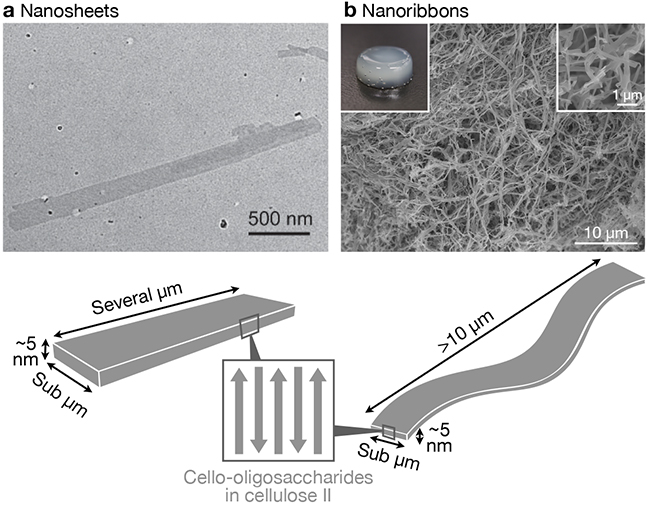
As a result of self-assembly of cello-oligosaccharides, nanosheet25,33 or nanoribbon16,34–40-shaped structures are produced (Figure 2). These nanostructures are lamellar crystals of cello-oligosaccharides in which elongated molecular chains align in antiparallel manner and have the thermodynamically most stable allomorph, cellulose II; cello-oligosaccharide chain terminals are located at the planes of the nanosheets or nanoribbons. Therefore, except for special cases where crystal allomorph changes18,41, the self-assembly of cello-oligosaccharides with terminal functional groups typically results in nanosheets or nanoribbons with functional groups displayed on the surfaces. Thus, the self-assembly of cello-oligosaccharides with terminal functional groups is a useful method for constructing functional nanostructures.
Paper has long been used in everyday life and recently attracted attention as a raw material for producing highly functional materials, including diagnostic and electronic devices. For such applications, paper is often modified with functional molecules (or nanoparticles). Physical modification, in which functional molecules are introduced noncovalently to surfaces of paper42–47, is simpler and generally requires less amount of environmentally harmful chemical reagents than chemical modification based on covalent bonding. However, the stability of physically modified state is not necessarily high, and the possibility of leakage of modified molecules tends to be relatively high. It is especially difficult to maintain a physically modified state in water because surfaces of paper are hydrophilic.
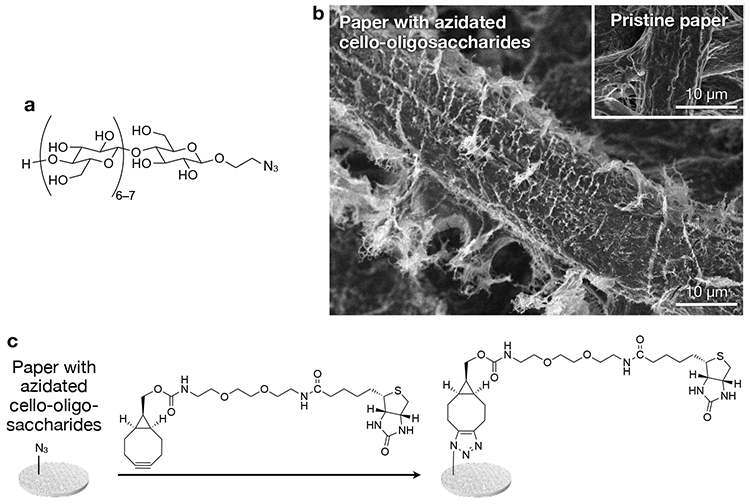
We have recently reported a novel physical modification method based on neutralization-induced self-assembly of cello-oligosaccharides with terminal functional groups28. This report used cello-oligosaccharides with azido groups (Figure 3a). Paper was infiltrated with aqueous alkaline solutions of azidated cello-oligosaccharides, followed by neutralization using hydrochloric acid. As a result, azidated cello-oligosaccharides self-assembled into hairy nanostructures that covered microfibers of paper (Figure 3b). Structural analyses suggested that cello-oligosaccharide chains constituting the hairy nanostructures were in cellulose II, which was different from cellulose I of paper microfibers. This difference in allomorph implies that cello-oligosaccharides self-assembled via heterogeneous nucleation on microfiber surfaces rather than via epitaxial crystal growth. The physically modified state of paper surfaces with azidated cello-oligosaccharides was stable in water for a long time. It is suspected that interactions between cello-oligosaccharides and cellulose is strong due to the identity in their chemical structure and responsible for high stability of the physically modified state.
Paper with self-assembled azidated cello-oligosaccharides has click (Huisgen cycloaddition) reactivity. As a proof of concept, biotin was introduced by the click reaction to the azidated paper (Figure 3c) for sensitive detection of anti-biotin immunoglobulin G28. Notably, detection sensitivity hardly decreased even after long-term storage of the modified paper, indicating a high stability of the physically modified state.
This report employed only azidated cello-oligosaccharides. Nevertheless, various terminally functionalized cello-oligosaccharides should be applicable to the physical modification for paper. Therefore, the self-assembly of functionalized cello-oligosaccharides is a promising method for simply and stably introducing functional groups into cellulose materials.
Conventional cellulose materials, such as paper and cloth, are composed of microfibers and have no significant nanostructures. On the other hand, studies have revealed that so-called nanocelluloses (e.g., cellulose nanofibers and nanocrystals) exhibit characteristic functional properties derived from their nanoscale structures for novel applications. Therefore, the development of technology for nanostructuring conventional microfibrous cellulose materials will greatly expand the versatility and usability of this sustainable biopolymer.
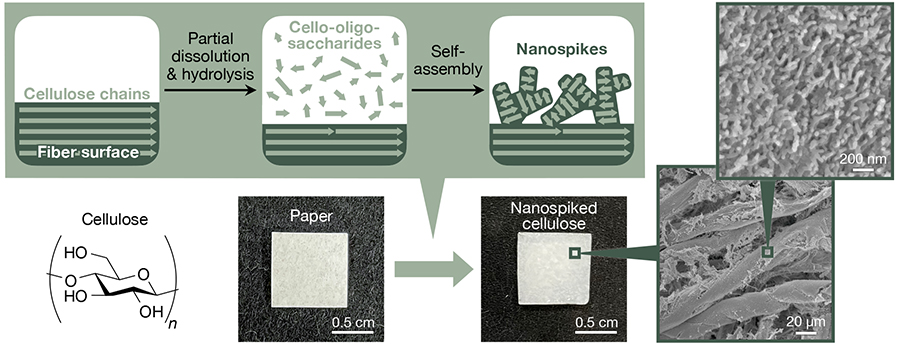
We have recently demonstrated the nanostructuring of conventional cellulose materials via partial dissolution and hydrolysis of cellulose for producing cello-oligosaccharides and their subsequent self-assembly into nanospike-like structures on microfiber surfaces (Figure 4)32. Filter paper with a high cellulose content was used in the study as a model cellulose material. Aqueous phosphoric acid solutions at 81% were applied to filter paper and left to stand at 45 °C for 20 h. As a result, partial dissolution and hydrolysis of cellulose occurred while maintaining the microfibrous structures of filter paper, generating cello-oligosaccharides with DPs of ~8. Then, water as coagulant was added to the phosphoric acid-swollen paper containing cello-oligosaccharides to induce the self-assembly of cello-oligosaccharides, resulting in the formation of spike-like nanostructures on microfiber surfaces. Infrared spectroscopy suggested that the cello-oligosaccharides constituting nanospikes were in cellulose II allomorph. Similar to the azidated cello-oligosaccharides described above, cello-oligosaccharides appeared to self-assemble via heterogeneous nucleation on microfiber surface for the formation of nanospike structures.
The resultant nanospiked cellulose was shown to have an ability to synthesize silver nanoparticles32. Heat treatment for nanospiked cellulose in aqueous silver nitrate solutions resulted in the synthesis of silver nanoparticles with particle sizes of several nanometers on the nanospiked surfaces. Analyses suggested that cello-oligosaccharides with relatively low DP in the nanospikes dissolved and acted as reducing agents during heat treatment, thereby reducing silver ions. In vitro tests showed that the resultant silver nanoparticle composites exhibited a high bactericidal activity. Additionally, our preliminary experiments suggested that cello-oligosaccharide nanospikes had significant effects on bacterial adhesion behavior. We are exploring the potential of nanospiked cellulose as biomedical materials.
Cello-oligosaccharide assemblies exhibit high thermal stability, biocompatibility, and insolubility in water and various organic solvents, similar to cellulose assemblies10. Moreover, the shorter molecular chain length prevents entanglement, providing better control over the self-assembly and consequent nanostructures of cello-oligosaccharides than cellulose. However, because of the lack of entanglement, macroscopic materials composed of cello-oligosaccharides alone are mechanically brittle. Although this brittleness is attractive for some applications such as cell culture scaffolds26, it can be a disadvantage in many applications. In this context, we believe that the self-assembly of cello-oligosaccharides for functionalizing and nanostructuring conventional cellulose materials described in this article is one of the great possibilities of cello-oligosaccharide assemblies in macroscopic materials. We would like to continue to explore the potential of cello-oligosaccharides for functionalizing ordinary materials.



