
Jun Hirabayashi
Tokai National Higher Education and Research System, Nagoya University, Japan. Ph.D, Science.
After graduated from Tohoku University (Master of Science), he started his professional carrier at Teikyo University under supervision of Prof. Kenichi Kasai for the investigation of animal lectins. On the occasion of GlycoXV (Tokyo, 1999), he proposed the concept glycome; for this realization, he moved to National Institute of Advanced Industrial Science and Technology (AIST, Tsukuba) in 2002, and was involved in a series of national projects for glycan engineering, while he was a deputy director in Research Center for Medical Glycoscience (2006~), and a prime senior researcher of Research Center for Stem Cell Engineering (2012~). Now, he is a designated professor in Institute for Glyco-core Research (iGCORE), Tokai National Higher Education and Research System, Nagoya University, as a project manager (HGP), while being a vice president of the Japanese Society of Carbohydrate Research (JSCR) and Japanese Consortium for Glycoscience and Glycotechnology. (JCGG). He is also a visiting professor of Kagawa University (2003~) and Yokohama City University (2019~).
In the preceding chapter (Part 1), the authors described the origin of galectins while focusing on the point that galectins are essentially cytosolic proteins1. In this chapter, one of the authors (J.H.) hypothesizes that the presence of carbohydrates is prerequisite to the generation of lectins that recognize them as counterpart molecules, and that the presence of galactose (the carbohydrate ligand of galectin) is prerequisite to the generation of galectin. Clearly, the question is: what is the origin of galactose? In this chapter and as previously proposed by Hirabayashi (for details, see refs. 1-4), a hypothesis regarding the “late arrival of galactose” and the origin of saccharides is presented.
Before thinking about the origin of galactose, let’s look around our natural environment where we find that basically all naturally occurring saccharides have the ᴅ-form (Note 1). Then ask: are all ᴅ-form monosaccharides found in nature? The answer is No. For example, the main aldohexoses in nature are ᴅ-Glc, ᴅ-Man, and ᴅ-Gal, while other aldohexoses are rarely found. ᴅ-Fru is the only discernible ketose. There are a number of isomers in ‘theory’, but that number is very small in actuality.
Note 1) The ʟ-saccharides are ʟ-Ara, ʟ-Fuc, and ʟ-IdoA. However, all of them are biosynthesized starting from ᴅ-saccharides, such as ᴅ-Glc and ᴅ-Man. The reason they are termed ‘L’ is that the hydroxyl group at the farthest chiral carbon (C4 or C5) is epimeric, i.e., facing left, in each of them.
Fig. 1 shows a series of ᴅ-aldoses. When comparing the structures in their 4C1 conformation, which is supposedly a stable chair form, we should note that all of the aldohexoses occurring in nature have no or a minimal number of, 1,3-diaxial interactions (Note 2), which is thought to destabilize the chair form structure. Namely, the number of 1,3-diaxial interactions is 0 for glucose, 1 for both mannose and galactose, and 2 or more for other monosaccharides. It is estimated that energy increase (destabilization) by inclusion of one 1,3-diaxial interaction is approximately 0.5 kcal/mol. We suggest that monosaccharide stability cannot be understood only in terms of the number of 1,3-diaxial interactions. However, it is possible to reach a similar conclusion by using other evaluation methods.
Note 2) As a most representative case, in cyclohexane, there is a steric interaction between the alternate axial substituents, i.e., between the C1 and C3 positions. The bulkier the substitution at the alternate position, the more significant will be the steric repulsion. Similarly, in pyranoses, there is a significant range of 1,3-diaxial interactions depending on the structures at the diastereomer level (e.g., glucose and mannose) and at the chair form level (i.e., 4C1 or 1C4). There is no significant 1,3-diaxial interaction in glucose in its 4C1 form, because it has no axial substituent at the C2, C3, C4, and C5 positions (i.e., all equatorial configurations). On the other hand, mannose and galactose have one axial substituent at C2-OH (mannose) and C4-OH (galactose), and the number of 1,3-diaxial interactions is modest. Of note, the number of 1,3-diaxial interactions in allose is two, albeit it has only one axial substituent (at C3) like mannose and galactose (which are all glucose epimers).
For example, aldehyde forms of aldoses, which can be measured using polarography, are present in different ratios. The ratio depends on equilibration between open-circular (aldehyde) forms and closed-circular (pyranose) forms. Generally, the latter form is much more stable and thus dominant (Table 1). In fact, the measured ratio of the aldehyde form of glucose is as low as 0.024%, and those of mannose and galactose are almost thrice that of glucose. Strange to say, the ratio of the aldehyde form of allose is considerably higher (1.38% or 58-times higher than the ratio of the aldehyde form of glucose), although allose has only one axial OH group like mannose and galactose. In fact, neither ᴅ- nor ʟ-allose occurs in nature.
| Aldose | Aldehyde form (%) | Relative to Glc | No. of 1,3-diaxial interactions |
|---|---|---|---|
| (Hexose) | |||
| Glucose | 0.024 | 1 | 0 |
| Mannose | 0.064 | 2.7 | 1 |
| Galactose | 0.082 | 3.4 | 1 |
| Allose | 1.38 | 58 | 2 |
| (Pentose) | |||
| Xylose | 0.17 | 7.0 | 0 |
| Arabinose | 0.28 | 11 | 1 |
| Lyxose | 0.40 | 17 | 1 |
| Ribose | 8.5 | 354 | 2 |
When the author started his professional career in the laboratory, a simple question was raised about lectin specificity. Because there were so many structures of monosaccharides with different names, he expected lectin specificities also to be diverse, and categorized into many types on the basis of different monosaccharides. However, it was not true. A series of primers and review articles made clear that the actual number of lectins specific for monosaccharides is rather small (Table 2). Probably, this observation applies to the present situation5. Indeed, we find papers reporting a number of lectins specific for mannose and galactose, but none reporting lectins specific for idose and altrose.
| Lectin families | Biological distribution | Monosaccharide specificities |
|---|---|---|
| Galectin | Animals | Gal |
| Legume lectin (L-type) | Plants, Animals | Glc, Man, Gal, Fuc, Sia |
| Ca-dependent lectin (C-type) | Animals | Man, Gal, Fuc (SLX) |
| Siglec (I-type) | Animals | Sia |
| Ricin B chain-like lectin (R-type) | Plants, Animals, Microorganisms | Gal, Sia |
| Fucose-specific lectin (F-type) | Plants, Animals, Microorganisms | Fuc |
| Hevein-like lectin | Plants, Animals | GlcNAc, Sia |
| GNA-like lectin | Plants, Animals | Man |
| Jacalin-like lectin | Plants, Fungi | Man, Gal |
| AAL-like lectin | Microorganisms | Fuc |
| Influenza virus (HA) | Viruses | Sia |
As a result of years of further consideration, the following two conclusions were obtained.
Note 3) Based on monosaccharide specificity, a Doyle6 classification of 237 lectins from animals (61), plants (154), and microorganisms (22) identified 145 (61%) Gal/GalNAc-specific lectins, 33 (14%) Glc/Man-specific lectins, 28 (12%) GlcNAc (chitooligosaccharide)-specific lectins, 16 (7%) ʟ-Fuc-specific lectins, and 15 (6%) Sia-specific lectins. Since then, the genome-wide search for new lectins and their functional analysis have much advanced, and thus, the above values may change to some degree. However, the dominance of galactose-specific lectins would not be expected to change, even though it is a fact that galactose-specific lectins can be more easily investigated by the presence of some useful tools for this purpose (asialofetuin or lactose-immobilized column).
On the first point, it is a priori true, and hence, regarded as a basic assumption.
The second point, if true, means that galactose has some unique features which other monosaccharides lack. As described in the previous chapter1, galactose has an advantage as a “recognition saccharide”. Possibly, because it worked well, this recognition system utilizing galactose developed variously. As a result, diverse lectins have been generated in the course of natural evolution. In fact, diverse protein families contain galactose-binding lectins, and their detailed galactose-binding mechanisms are significantly different1,7. Regarding galectins, the fine specificities of individual galectins for glycans are also divergent8,9.
Theoretically, the number of aldohexoses is 16. However, the number of monosaccharides we actually find in nature is only 3, that is, ᴅ-Glc, ᴅ-Man, and ᴅ-Gal. Consider the following question: do these monosaccharides have some relationship from a biosynthetic viewpoint?
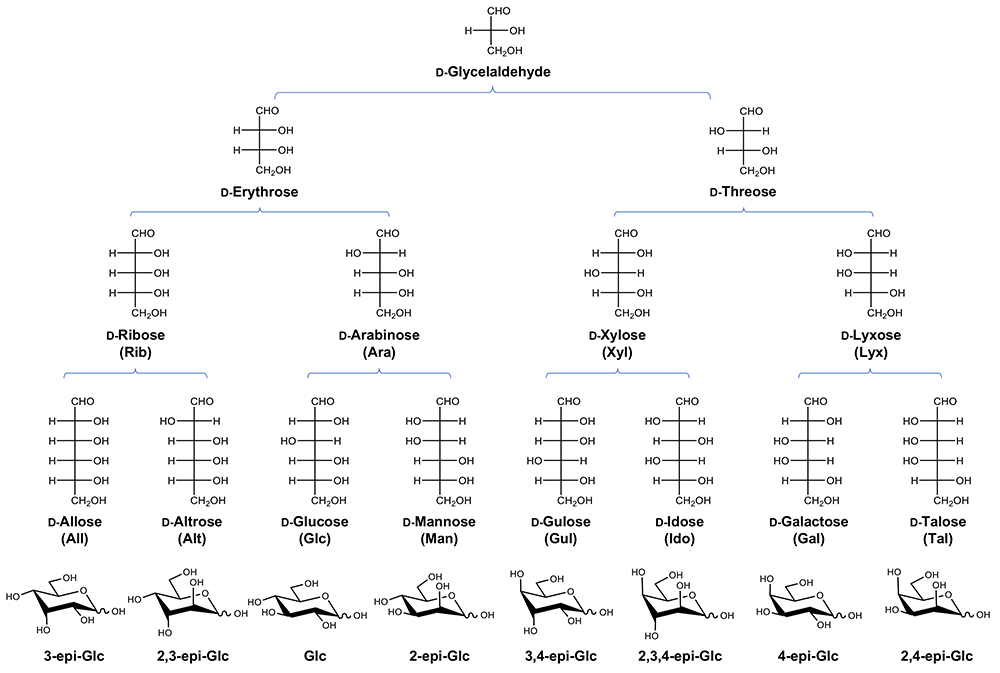
To begin with, ᴅ-Glc and ᴅ-Man are epimers differing in the configuration of the hydroxyl group at the C2 position (C2 epimer). Being a neighbor to the C1 aldehyde group, C2 is a chemically specific position. In fact, it has been long known that ᴅ-Glc is converted to ᴅ-Fru and ᴅ-Man under alkaline conditions (Fig. 2). Named after its discoverers, the reaction is known as the Lobry de Bruyn – Alberda van Ekenstein transformation10,11. This reaction proceeds ‘chemically’ in the absence of enzyme, and indeed, it is utilized for industrial production of some ketoses. Another important point is that the reaction reaches equilibrium in the presence of an appropriate basic catalyst, and thus, ratios of the reaction products (a mixture of ᴅ-Glc, ᴅ-Fru, and ᴅ-Man) are theoretically the same regardless of the ratio of the starting materials. In contrast, almost all organisms are equipped with specific enzymes to catalyze these reactions.
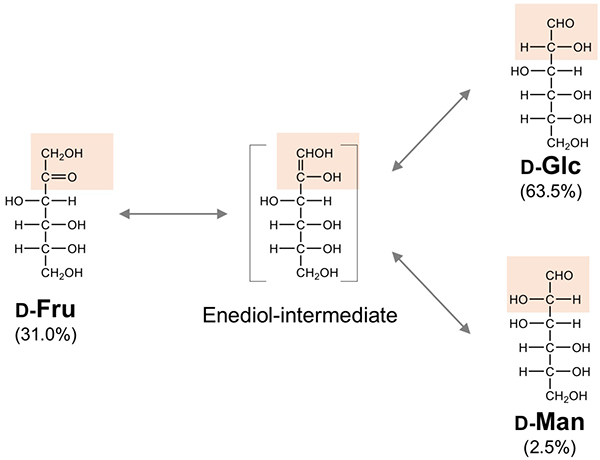
On the other hand, galactose is also biosynthesized from glucose, but the features of this biosynthesis are considerably different: first, the reaction center is not C1 (aldehyde), but C4 at the opposite end of the molecule. It is necessary to convert C4-OH (4-epimerization) to make galactose from glucose. The enzyme named UDP-glucose-4-epimerase is responsible for this reaction, which oxidizes C4 converting the sugar to a 4-keto-intermediate with the help of NAD+, then the enzyme adds back the removed hydrogen to the opposite side of C4 to convert it to a C4-OH group and complete the C4 epimerization.
Second, the pyranose ring is not open and remains in the closed state for the reaction to proceed (Note 4). Third, this epimerization is enabled by a redox reaction utilizing a coenzyme, NAD+. Interestingly, similar reactions via a 4-keto-intermediate are widely found not only in galactose biosynthesis, but also in conversion to many other saccharides and derivatives. Such a feature making maximal use of a few reaction mechanisms to produce various other things from abundant, low-cost materials is called ‘bricolage’ (Note 5).
Note 4) Of note, in the above Lobry de Bruyn transformation, ring opening is mandatory, whereby the aldehyde group at C1 is subjected to isomerization via keto-enol tautomerization between C1 and C2.
Note 5) This word was created by a French cultural anthropologist, Claude Lévi-Strauss (1908-2008). He described the characteristic patterns of thinking that people used in undeveloped countries to create something new from combinations of materials at hand, which is in contrast to the thoughtful engineering approach currently used to design and manufacture products in modern countries.
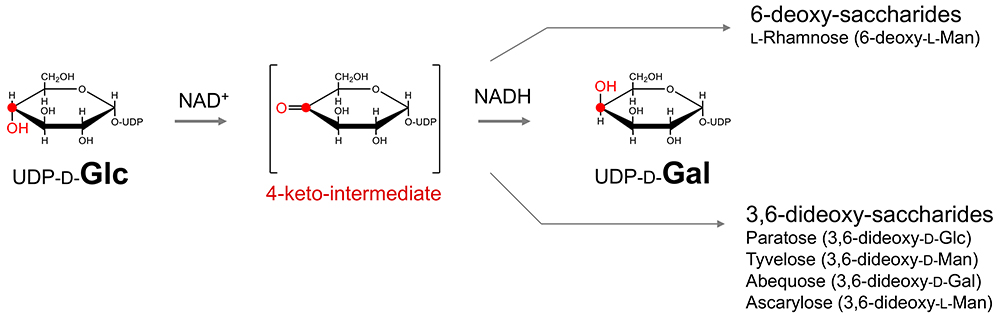
When considering biological evolution, introduction of the 4-keto intermediate is strategically important, because it enables modification at not only the C4 position, but also the ‘chemistry’ around it. For instance, the keto-enol tautomerization reaction between the C4 and C5 positions enables not only 5-epimerization, but also 6-deoxidation associated with this event. In fact, 6-deoxy-saccharides, such as ʟ-fucose and ʟ-rhamnose, are biosynthesized via transformation of some corresponding 4-keto-intermediates.
The C3 position plays a similar role. In fact, 3,6-dideoxy-saccharides, e.g., paratose (3,6-dideoxy-ᴅ-Glc), abequose (3,6-dideoxy-ᴅ-Gal), colitose (3,6-dideoxy-ʟ-Gal), tyvelose (3,6-dideoxy-ᴅ-Man), and ascarylose (3,6-dideoxy-ʟ-Man), are all biosynthesized via transformation of some 4-keto forms used as key intermediates. To summarize, the aldehyde group at the C1 position governs the C2 position [C1 strategy], while the C4 position governs epimerization and modification at C4 and all other positions (C3 and C5) [C4 strategy]. Thus, the feature fits the Ockham’s razor principle perfectly (Note 6).
Note 6) An English philosopher in the 14th Century, Ockham introduced the principle known as “the principle of parsimony” or the principle that “entities should not be multiplied beyond necessity”. The razor here symbolizes the act of shaving or cutting away unnecessary items.
The two described isomerization patterns are compared in Table 3. It should be mentioned that they utilize a common mechanism to convert the carbonyl carbon (sp2) to chiral sp3.
| 1. Alkaline-based isomerization analogous to the Lobry de Bruyn transformation [C1 strategy] |
|---|
|
| 2. Isomerization via C4 keto (oxidized) intermediate [C4 strategy] |
|
Among glycans, N-glycans are the most characterized type, as their biosynthetic mechanisms are well studied. Indeed, they are conserved among eukaryotes and share features including a common precursor for their biosynthesis, although it is difficult to explain why a structure like Glc3Man9GlcNAc2 became the common precursor in eukaryotes. More confusing to beginning researchers in the field is that while N-glycans are biosynthesized from this common precursor, it is further subjected to an elaborate process of maturation from high-mannose-type structures to more complex type structures. Most of the first 9 mannose residues are removed in this process, and a lactosamine structure (Galβ1-4GlcNAc) with variable branching patterns is subsequently attached, which are further modified by ʟ-Fuc and sialic acids (e.g., Neu5Ac and Neu5Gc). This results in a transformation into heterogeneous structures distinguishable by various modifications12. It is quite difficult to understand why such an effort has to be made. The above-described maturation process occurs largely in mammals including humans, while unique maturation processes have been developed in other organisms (evolution observed in individual species). Nevertheless, the component monosaccharides used for N-glycans remain totally the same.
Regarding the N-glycan precursor, like a tough nut it is hard to crack. Notably, the chitobiose structure (GlcNAcβ1-4GlcNAcβ-Asn) occurs in the reducing terminus. Here, we present a bold hypothesis: component monosaccharides should have appeared in a stepwise evolutionary manner. That is, the earlier the monosaccharides appeared in evolution, the closer they would be located to the reducing terminus. This idea is essentially the biological rule called “terminal addition” 13-15. This experimental rule is based on the observation that a new characteristic of an organism is always added to old ones.
Glycans are biosynthesized in the endoplasmic reticulum and Golgi apparatus, which contain hundreds of glycosyltransferases that transfer a series of monosaccharides in a step-by-step manner to construct a series of glycans. The author’s belief is that component monosaccharides are likely to be structurally integrated in the order of their evolutionary appearance. For example, probably the oldest members like ᴅ-Glc and ᴅ-GlcNAc were incorporated in the glycan structure at a very early stage of evolution, and thus, found nearest to the reducing terminus. In the next stage, ᴅ-Man was incorporated as a result of Lobry de Bruyn rearrangement using ᴅ-Glc, and finally, ᴅ-Gal, a newcomer saccharide. This is an outline of the “galactose late-comer” hypothesis proposed by the author2-4.
By the way, reducing terminal location of chitobiose is meaningful, considering that chitin is the oldest polysaccharide. Chitin consists of β1-4-linked GlcNAc to make a linear structure, and has essentially the same backbone structure as that of peptidoglycans (GlcNAcβ1-4MurNAcβ1-4), which are common to all bacteria (Note 7).
Note 7) MurNAc is a GlcNAc derivative modified by lactate at the C3 position and is used to link a pentapeptide; therefore, it has the same peptidoglycan backbone structure as chitin. Of note, however, bacterial biosynthesis of peptidoglycan is performed by bacterial transferases responsible for GlcNAc and MurNAc, and thus, chitin is not used as a precursor. On the origin of GlcNAc, it is possible that glucose was used to detoxify undesired ammonium generated as an inevitable result of metabolism, whereas contemporary organisms utilize glutamine as an ammonium source for GlcNAc.
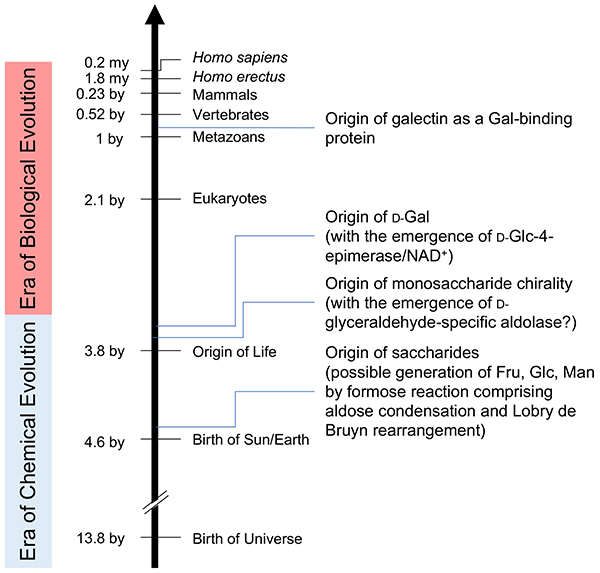
As described in the previous chapter1, assuming that all galactose-binding lectins including galectins first appeared 1 billion years ago, when metazoans emerged, galactose must have existed at that time, and some primitive organisms may have synthesized and utilized it. “Why β-galactoside” is another unsolved question, but if we take it into consideration that the present biosynthesis is somewhat complicated requiring the generation of an NAD+-mediated 4-keto intermediate, the origin of galactose must not be very old, and was at the earliest after the first organism appeared on earth (3.8 billion years ago or later).
On the other hand, glucose can be generated by a chemical reaction involving Lobry de Bruyn transformation, and the formose reaction, a classic glycochemical reaction, is essential to explain how carbohydrates are produced from the simplest organic unit defining their structure, i.e., formaldehyde (HCOH), possibly present in the primitive atmosphere. Actually, a variety of carbohydrates are generated by this reaction via the trioses glyceraldehyde and dihydroxyacetone, which in turn are used to produce ketoses (e.g., fructose) by aldol condensation (Fig. 4). If fructose undergoes a Lobry de Bruyn transformation, it is easily converted to glucose as described. Considering that saccharides are generally reductive, and also reactive with the amino groups of amino acids and peptides, their stability is anticipated on the primitive earth. However, glucose is the most stable monosaccharide, and hence, it seems probable that glucose accumulated on the primitive earth in the era of chemical evolution, i.e., before life began, and was utilized by the first life forms.
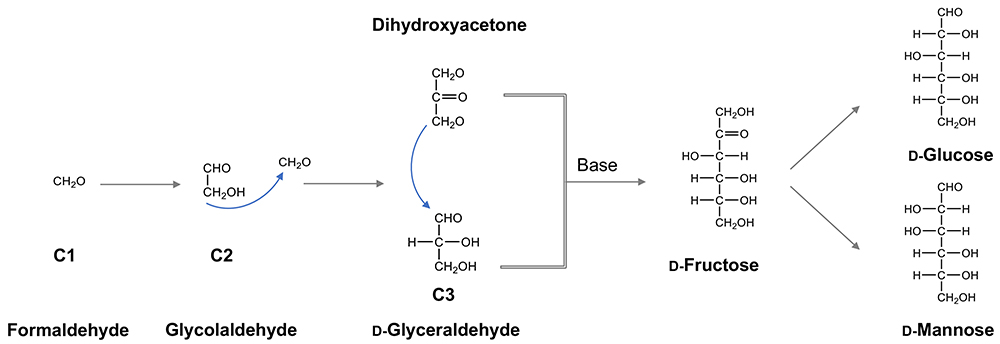
Lastly, the author would like to mention a little bit about the origin of chirality. It is highly possible that the first monosaccharides on the primitive earth were racemic mixtures of both ᴅ- and ʟ-forms. However, what happens if in the first primitive life form a primitive enzyme is created that could catalyze the aldol condensation reaction (to generate hexoses including fructose from glyceraldehyde and dihydroxyacetone)? Here, this primitive enzyme is considered to have preferred either ᴅ- or ʟ-glyceraldehyde, whereby all the monosaccharides generated in the following processes should preserve that chirality (Scheme 1). Namely, the origin of ᴅ-enantiomerism is attributed to the ᴅ-selectivity which the primitive aldolase of the first life form happened to develop.
ᴅ-Glyceraldehyde + Dihydroxyacetone → ᴅ-Fru → ᴅ-Glc → ᴅ-Gal
ʟ-Glyceraldehyde + Dihydroxyacetone → ʟ-Fru → ʟ-Glc → ʟ-Gal
Scheme 1. Chirality preserved by the formose reactionIn the defined formose reaction mechanism, only ᴅ-series saccharides are produced from a ᴅ-saccharide, and vice versa.The origin of galectins which acquired galactose-binding ability is estimated to be a billion years ago or later, and probably in the era when a variety of metazoans first arose. The origin of ᴅ-Gal, which is assumed to have been substantially produced by using NAD+ as a coenzyme and ᴅ-Glc, is estimated to be 3.8 billion years ago or later. The origin of glucose is in the era of chemical evolution (3.8 billion years ago or earlier) and must have occurred before that of ᴅ-Glc (i.e., 3.8 billion years ago or later). From the viewpoint of prebiotic synthesis of saccharides, i.e., on the basis of formose reactions including Lobry de Bruyn transformation, the first fructose must have arisen a fraction of a second earlier than the first glucose. An overall picture of the evolutionary relationship of galectin and galactose, as well as related issues, is shown in Fig. 5.
The author deeply thanks Dr. Jun Iwaki (Tokyo Chemical Industry Co. Ltd., Tokyo) and Dr. Ryuichiro Suzuki (Akita Prefectural University, Akita), who contributed to the preparation of figures and joined the discussion. He also thanks Dr. Kenichi Kasai (a professor emeritus at Teikyo University, Tokyo), who helped prepare the discussion of the origin of monosaccharides including galactose. Dr. Sachiko Sato (Laval University, Quebec City) is also acknowledged for her continuous commitment to the editing.



