
Tomoya Imai
Associate Professor, Research Institute for Sustainable Humanosphere, Kyoto University
Doctorate (Agriculture), Kyoto University, 2000
Research Associate, Tokyo University of Agriculture and Technology, from April 2000
Post-doctoral fellow, CNRS-CERMAV (Centre de Recherches sur les Macromolécules Végétales), from October 2000
Post-doctoral fellow, Graduate School of Science, Kyoto University, from April 2002
Current position from April 2008
My research focuses on the synthetic biology of plant cell walls through a combination of electron microscopy and X-ray structural analysis with the biochemistry of membrane proteins.
Cellulose is a natural polysaccharide, and is functionally classified as a structural polysaccharide. Its superior strength is attributed to the fact that it is composed of multiple molecular chains, and it has a structure known as cellulose I crystal that exhibits a high crystalline modulus. The fact that such an agglomeration of polymers can be synthesized by enzymatic proteins suggests that cellulose biosynthesis is a mechanism designed to synthesize the strongest possible structure by controlling the polymer chains at ambient temperature and pressure in aqueous solvents. In comparison with the typical formation process for general-purpose polymers, which involves high temperature, high pressure, and harsh solvents, the enzyme cellulose synthase possesses an extremely sophisticated “green” mechanism for controlling polymer structure.
In this paper, I will describe efforts to reconstitute the cellulose-synthesizing activity of cellulose synthase, the mechanism of which we have been seeking to elucidate for more than 10 years.
Natural polysaccharides are glycopolymers synthesized by living organisms. On a molecular scale, they are described as polymers that are synthesized by enzymes. Enzymes are proteins (some are RNA molecules) with catalytic functions and are characterized by their high specificity for particular reactions. The biosynthesis of natural polysaccharides such as pectin and gum arabic, with their diverse constituent monosaccharides and binding modes, is an example of molecular synthesis in which the functions of enzymes are on full display.
Cellulose, on the other hand, has a simple molecular structure, consisting of a single type of monosaccharide (glucose) connected by one type of glycosidic linkage (β1→4-glycosidic linkage), and its synthesis is a simple function in terms of reaction specificity. However, given that cellulose is a polymer that is insoluble in water and is synthesized in a crystalline fiber structure consisting of multiple molecular chains, we can see that the function of cellulose synthase is in fact extremely sophisticated. To more fully understand this phenomenon, we will discuss the higher-order structure of cellulose in the next section.
Structural polysaccharides such as cellulose are polysaccharides synthesized by living organisms for their own protection. Cellulose is the main component of the cell walls of tall trees tens of meters high, and chitin is the main component of the exoskeletons of crabs and insects. Polymer materials that function this way generally do not exist as single molecules, but rather as agglomerates of multiple molecular chains (Fig. 1). High-strength polymers generally exhibit highly regular molecular assemblages and crystallinity, but cellulose also exists in a crystalline fiber structure in which multiple molecular chains are agglomerated.
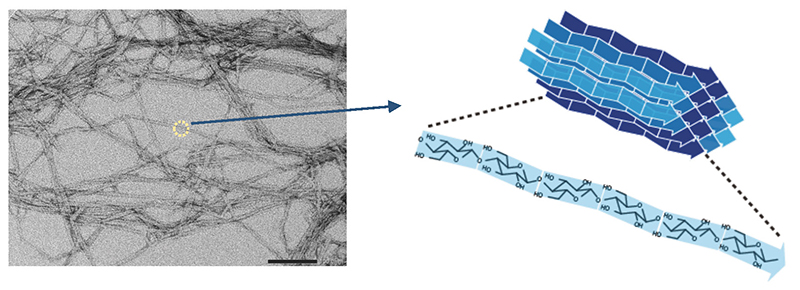
The fact that such a material as cellulose is made by living things is certainly an impressive achievement. Although there are multiple possibilities for the crystal structure of cellulose (known as crystal polymorphism), it is an observed fact that natural cellulose, without exception, exhibits only one type of crystal structure known as cellulose I 1,2 (Cellulose I occurs as two subtypes, Iα and Iβ3,4; in this paper, they will be collectively referred to as type I, unless otherwise specified). Moreover, cellulose I has the highest tensile strength (crystalline elastic modulus) of all the crystal polymorphs of cellulose5 (Table 1). In other words, living organisms fully exploit the potential of the cellulose material they make.
| Crystal polymorph | Elastic modulus, GPa (tensile) |
| I(Iβ) | 138 | II | 88 | IIII | 87 | IIIII | 58 | IVI | 75 |
A question that arises here is whether the fiber structure of natural cellulose in the form of cellulose I crystals is the result of a spontaneous process due to the molecular chain properties of cellulose or an active mechanism? The answer to this question appears to be the latter. Although many researchers have described efforts to artificially produce cellulose by enzymatic synthesis6-9, simple synthetic systems almost always result in the formation of laminar crystals in which oligomers are agglomerated, and which have the energetically most stable type II crystal structure. These artificial enzymatic synthesis systems are characterized by joining cellulose with β1→4-linkages via elaborate chemical reaction designs, and the synthesized cellulose molecules liberated into aqueous solvents aggregate to form the energetically most stable type II crystal structure in most instances.
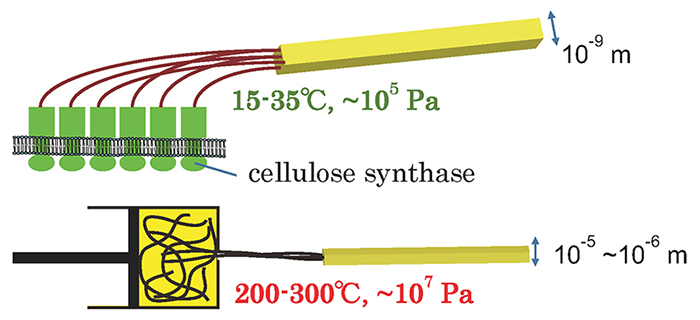
In summary, the cellulose synthase protein found in living organisms has the ability to synthesize high-molecular-weight cellulose (polymerization function) and the ability to bundle the high-molecular-weight cellulose in a regular manner and synthesize it as the strongest type I crystal (crystallization function) with a fiber structure. In other words, cellulose synthase can assemble cellulose polymers without applying heat or pressure in water, a poor solvent, and synthesize them into a specific structure (the fiber structure of type I crystals).
Compared to the synthesis conditions required to control the structure of various polymers such as polypropylene, this function is astonishing, and can therefore be regarded as a “green” mechanism for controlling polymer structure (Fig. 2).
Hence, the conventional biochemical analysis and concepts that are usually used in enzyme research are not sufficient to shed light on the function of cellulose synthase; polymer science analysis and concepts are also important. In this paper, I will outline our attempts to elucidate this superb technology possessed by living organism.
The basic approach in reconstituting enzyme activity involves extracting the enzyme protein and initiating its activity in an in vitro environment. The first step in this process is the selection of a model organism with which to conduct the experiment. In the case of cellulose synthase, the cellulose-producing bacterium Acetobacter has been studied as a model organism since the 1950s10, and the process that led to the discovery of the cellulose synthase gene over the next 30 years11,12 is described in my 2011 paper13. At that time, identification of the gene was of the utmost concern, so there are few reports investigating the structure of cellulose synthesized by isolated enzyme activity; however, the crystal structure of the cellulose synthesized by the isolated enzyme was not the natural type I crystal but the energetically most stable type II crystal14. In other words, although the synthesis of cellulose was a success, the synthesized cellulose chains were aggregated into different solid structure from the native one. Although we should probably not overlook this enzyme research, gene identification11,12 and protein structure determination15 have taken precedence, and functional analysis is yet to catch up, even in a simple system such as bacteria.
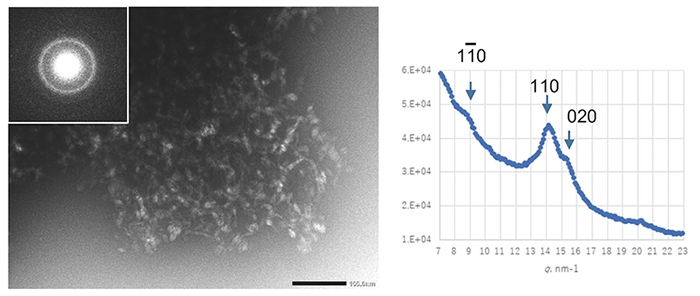
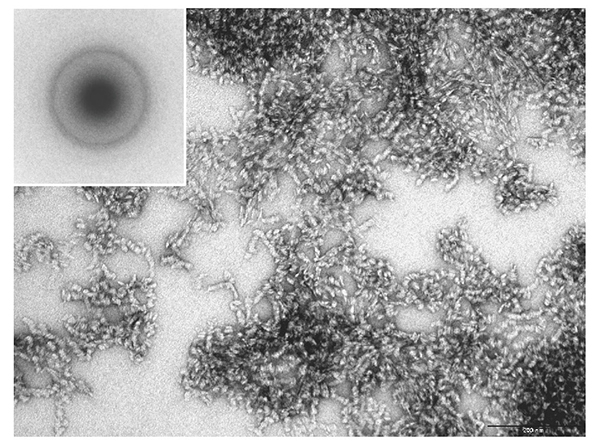
Cellulose synthase is a transmembrane protein embedded in the cell membrane and is extracted using a detergent for a biochemical assay. The number of detergents used in membrane protein research has increased dramatically since the 1980s, when cellulose-synthesizing activity was first extracted. By selecting a detergent that has been successful in structural biology research where protein stability is particularly required, an enzyme was extracted from Acetobacter cells under milder conditions and an attempt was made to reconstitute the natural activity in vitro16. Unfortunately, however, the synthesized cellulose had a type II crystal structure and a clump-like structure (Fig. 3).
Obtaining cellulose-synthesizing activity while maintaining natural activity failed, as in the earlier report, but the cellulose synthesized in this system was a high-molecular weight polymer with a weight-average degree of polymerization (DP) of about 30017. Therefore, it was confirmed that the functional deficit was only in the crystallization function of the second step and not in the polymerization function of the first step17, although DP was lower than that for original bacterial cellulose18. In order to redress this functional deficiency (partial denaturation), we tried screening for temperature conditions and synthesis reactions in a centrifuge (synthesis reaction under shear stress), but none of them could rescue the functional deficiency, and cellulose II-type clumped agglomerates were formed17.
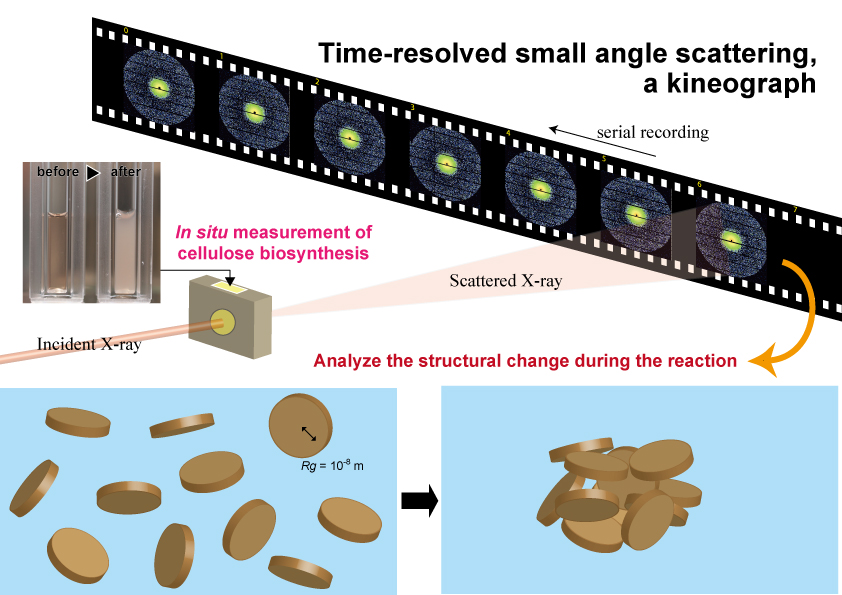
While experiments to recover from this partial denaturation in an in vitro system are still underway, it is important to correctly understand the behavior of cellulose molecular chains synthesized in a state in which this crystallization function is deficient. In this in vitro reaction, it was observed that the reaction solution became turbid as the reaction progressed (Fig. 4). This means that a structure with a size equivalent to the wavelength of visible light is formed as the reaction progresses. Therefore, we hypothesized that we could visualize the agglomeration process of cellulose molecules with higher resolution by using X-rays, which have a shorter wavelength than visible light, and conducted experimental research using the small-angle X-ray scattering technique19.
Since the scattering signals from molecules in water are extremely weak, strong X-rays from a synchrotron radiation facility are required to observe the progress of the reaction at reasonable time intervals. Therefore, we conducted time-resolved, small-angle X-ray scattering measurements using the BL40B2 beamline at SPring-8, capturing small-angle scattering patterns at 5-minute intervals while the synthesis reaction was carried out in a liquid cell (Fig. 4). The formation of the agglomerated structure was found to be a two-step process: first, an elementary structure with a radius of gyration (Rg) of less than 10 nm was generated, and after 15 minutes, when the structure exceeded a certain parameter (concentration or degree of polymerization, we assume), it formed a large, agglomerate structure (Rg of less than 30 nm). We speculate that the structure with a Rg of less than 10 nm formed in this first stage is one of the poorly agglomerated structures caused by the loss of crystallization function. This is an experiment that we hope to attempt in the future when we can reconstitute the native activity of cellulose synthase in an in vitro synthesis system.
The success of this experiment shows that small-angle X-ray scattering is a useful tool for visualizing the process of formation of not only cellulose but also structural polysaccharides and biomass in general. We hope to conduct similar research on various polysaccharides and biomass constituents and accumulate knowledge to explain the mechanism of biomass formation in general through comparison.
Next, in a different approach from the in vitro system, we decided to try a synthetic biology approach in which cellulose synthase is expressed in living cells and the cells then synthesize cellulose. The advantage of this approach is that the cellulose synthesis reaction can be reconstituted in a living cell, that is, with a cell membrane with potential differences between the interior and exterior of the cell. For this purpose, it is necessary to select genes that are necessary for cellulose synthesis and should be introduced into the cell.
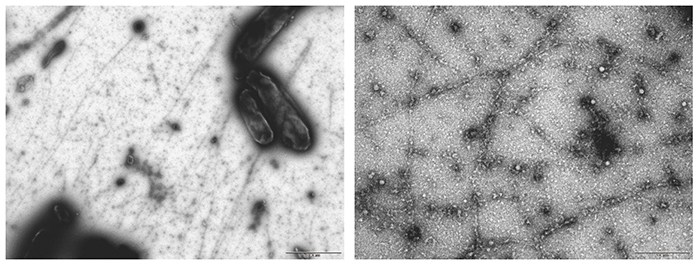
In Acetobacter, an operon consisting of four genes, cesA, cesB, cesC, cesD, has been identified as a cluster of cellulose synthase genes11. Among the proteins encoded by these genes, CesA (catalytic subunit) and CesB (auxiliary subunit) are known to be the minimum required for cellulose synthesis activity11,20; hence, we sought to synthesize cellulose in Escherichia coli (E. coli) cells by introducing the genes cesA and cesB into an E. coli cell line21. This experiment proceeded surprisingly well, and it was very exciting when I saw the white residue produced after treatment with acetic acid and nitric acid (so-called Updegraff treatment22) in the first trial. Structural analysis however showed that the synthesized cellulose was a clumpy agglomerate of type II crystals, showing a failure in the crystallization function as in the in vitro system (Fig. 5). On the other hand, the weight-average DP of the synthesized cellulose was over 700, indicating that the polymerization function was retained.
In conclusion, we have successfully reconstituted cellulose synthase activity in living E. coli cells but found that some factors required to synthesize the fiber structure of natural cellulose type I crystals are still missing. As mentioned above, the cellulose synthase complex contains four subunits, A, B, C, and D, as well as a carbohydrate-hydrolyzing enzyme encoded upstream of the cesABCD four-gene operon and a protein of unknown function called ccp, which have also been shown to be involved in cellulose-synthesizing activity23,24. These were expressed together with CesAB in E. coli. However, no matter what, agglomerated cellulose with type II crystals was still synthesized, indicating that it is not enough to simply express the necessary proteins.
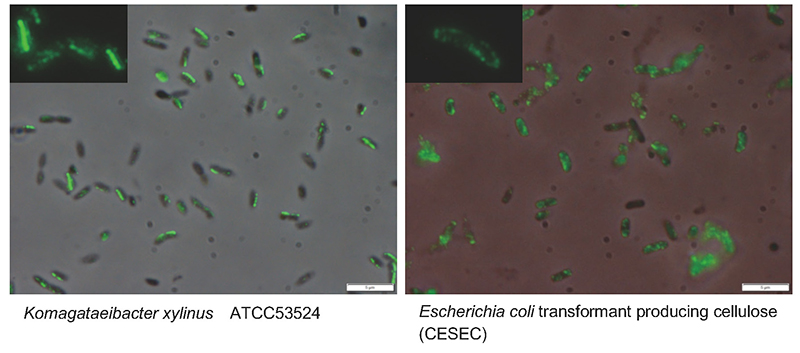
So, what other important factors are there? A promising candidate is the higher-order structure of cellulose synthase. On electron microscopy, cellulose synthase is observed to have an orderly granular structure on the cell membrane of cellulose-producing organisms25,26. This structure, known as the terminal complex (TC) because it is observed at the ends of cellulose fibers during synthesis, shows various morphologies depending on the species and is known to correlate with the structural features of the cellulose being synthesized27. Therefore, the regular arrangement of enzyme proteins is considered to be a necessary factor for the formation of cellulose I fiber structure in cellulose biosynthesis. In the case of Acetobacter, TCs are aligned along the long axis of the cell, and four molecules, CesA28 (Fig. 6, Left), CesB26, CesD24,28, and ccp24, have been shown to localize to TCs. However, when we examined the localization of CesA protein expressed in the E. coli system by immunofluorescence staining, it was found to be distributed throughout the cell (Fig. 6, Right). In other words, although cellulose synthase could be expressed on the cell membrane with polymerization function, it was not able to maintain the crystallization function because it was present in a disordered state on the cell membrane.
Therefore, the crystallization function in Acetobacter appears to be realized by the existence of an active mechanism to arrange cellulose synthases linearly along the long axis of the cell, as described in the discussion of the original work29. In fact, electron cryotomography, which has recently been used extensively in structural biology research, showed a cytoskeleton-like structure immediately below the cell membrane of the cellulose-synthesizing site30 and suggested that this may be a factor in the orderly arrangement of the cellulose synthase complex. These results suggest that a mechanism that controls the arrangement of cellulose synthase is the most plausible hypothesis for the crystallization mechanism.
The cellulose synthesized in this cell line showed a fiber structure when observed under electron microscopy before washing (Fig. 7). When we first saw these fibers under the electron microscope, we, before observing the washed product in Fig. 5, were excited that we had finally succeeded in reconstituting the native activity. However, in the subsequent washing process, the remaining sample turned out to be a clumpy agglomerate of cellulose type II crystals, as shown in Fig. 5. The process of converting cellulose I into cellulose II is known as mercerization, which involves treatment under strong alkaline conditions31. However, since the washing treatment of cellulose used in the above experiment was under extremely mild conditions, the fiber structure observed before washing could not be cellulose in the native structure. Thinking positively, we imagine that the fiber structure is a single molecular chain of cellulose, which was transformed into an energetically stable agglomeration of cellulose type II crystals by a much milder treatment than mercerization. If this is true, the synthetic biology approach, which allows us to encounter structures that we cannot encounter by observing natural samples, may play a crucial role in the fundamental study of biomass formation.
In this paper, I have introduced the procedures and results of synthetic biology research into cellulose synthase. Unfortunately, we are yet to reconstitute the natural activity of cellulose synthase. From a positive perspective though, we can convincingly explain that the key factor is the nature of the crystallizing function, if we can redress the functional deficiency. Alternatively, it is also possible that the crystallization function of cellulose synthase may be too sophisticated to be easily reproduced.
The control of macromolecular structure in aqueous solvents at ambient temperature and pressure, as emphasized in this paper, is a common feature in the synthesis of not only cellulose but also structural polysaccharides in general such as chitin, and biomass such as wood. Both in terms of biochemistry and macromolecular science, this is an intriguing and sophisticated function. Structural biologists are also beginning to explore the field of enzymatic proteins involved in biomass formation, such as cellulose synthases15,32. Research into biomass formation is expected to gain momentum in the future. Focusing on the higher-order structure of the synthesized biomass, we wish to make every effort to develop a multidisciplinary understanding of the life science of biomaterial synthesis.
Acknowledgments
The data presented in this paper are drawn from research conducted at the Research Institute for Sustainable Humanosphere at Kyoto University, and represent the contributions of many people. In particular, I would like to express my gratitude to Mr. Akira Hashimoto (currently at Rengo Co., Ltd.; similarly below), Dr. Yoshiki Horikawa (Tokyo University of Agriculture and Technology), Mr. Kenji Shimono (Azumi Filter Paper Co., Ltd.), Dr. Shijing Sun (Nanjing Forestry University), Dr. Paavo Penttilä (Aalto University), Dr. Satoshi Kimura (University of Tokyo), Dr. Hirotaka Tajima (Hosei University), and Professor Junji Sugiyama (Graduate School of Agriculture, Kyoto University). We also thank Grant-in-Aid for Scientific Research (Kakenhi) and JST-CREST for research support.



