1 Department of Pathology and Laboratory Medicine, University of California, Irvine, USA 2 Department of Neurology and Institute for Immunology, University of California, Irvine, CA 92697, USA. 3 Lunenfeld Tanenbaum Research Institute, Mount Sinai Hospital, 600 University Avenue R988, Toronto, Ontario, Canada M5G 1X5 4 Department of Molecular Genetics, & Department of Laboratory Medicine and Pathology, University of Toronto, Canada * Corresponding authors: James W. Dennis: DENNIS@lunenfeld.ca Michael Demetriou: mdemetri@uci.edu

Haik Mkhikian
Dr. Haik Mkhikian received a BA in Philosophy and BS in Biochemistry and Molecular Biology at University of California, Irvine in 2005. He then spent a year as a UGSP scholar at the NIH in Bethesda, MD before returning to UC Irvine for MSTP training. He completed my graduate training in Dr. Michael Demetriou’s lab studying the regulation of N-glycan branching in T cells, and its dysregulation in multiple sclerosis. After completing my MD/PhD training in 2016, he remained at UC Irvine as a research track Clinical Pathology resident, where he is currently a research fellow.

Michael Sy
Dr. Michael Sy is Assistant Professor in the Department of Neurology at the University of California, Irvine and the Co-director of the Multiple Sclerosis Regional Center at the Long Beach VA. He graduated from Yale University (2002) and complete his MD and PhD (2011) with the UC Irvine Medical Scientist Training Program. Residency training in Neurology and fellowship in Neuroimmunology (2016) at UC Irvine Medical Center. In addition to providing complete and holistic patient care, He is involved in basic, translational, and clinical research on myelination in Multiple Sclerosis.

James Dennis
Dr. James Dennis received his BSc in mathematics at Concordia University, Montreal, PhD in Biochemistry at Queen’s University Kingston (1978), postdoctoral training at German Cancer Research Center (DKFZ), Heidelberg, and then was awarded a Terry Fox fellowship to work with Dr. Harry Schachter at University of Toronto (1980). He began as Assistant Professor at Queens University (1982), and moved to became a founding member of the Lunenfeld-Tanenbaum Research Institute (1985) and Professor in Molecular Genetic Department at the University of Toronto. He is a fellow of the Royal Society of Canada, held a Canada Research Chair in Glycobiology, inventor on 20 patents, and more than 200 publications, 20,000 citations, and importantly, mentored to many talented students.

Michael Demetriou
Dr. Michael Demetriou received his combined M.D. and Ph.D. (1996) at the University of Toronto, the latter in Molecular Genetics under the supervision of Dr James Dennis. He then completed a residency in Neurology at the University of Toronto, post-doctoral training in glycobiology with Dr James Dennis at the Lunenfeld-Tanenbaum Research Institute (2001) and clinical fellowship in Neuroimmunology with Dr Stanley van den Noort at University of California, Irvine (2002). Dr Demetriou became faculty at the University of California, Irvine in 2001 and is currently a clinician-scientist and Professor of Neurology, Microbiology and Molecular Genetics. His research has been continuously funded by the NIH for 20 years, with 44 publications and more than 5000 citations. He has served as a chartered member of two NIH study sections as well as mentoring nine PhD’s and numerous post-docs and undergraduate students.
The galectin lattice is a multi-valent interaction of galectins with glycoproteins at the cell surface that displays rapid exchange of binding partners with properties of liquid-liquid phase transitions, thereby acting as an intermediary between freely diffusing glycoproteins and stable complexes in the membrane. The galectin lattice (i) regulates flow of receptors and solute transporters to coated-pit endocytosis and/or caveolin domains, and (ii) promotes turnover of cell-cell contacts such as immune synapses and focal adhesion complexes. Metabolic regulation of UDP-GlcNAc supply to Golgi N-glycan remodeling regulates glycoprotein affinities for galectins –and in turn, trafficking and presentation at the cell surface. The lattice model has been validated in immune regulation, cancer progression and glucose homeostasis in mice. Here we review the interactions of metabolism, galectins and glycoprotein ligands as well as the utility of this model to predict and treat inflammation and autoimmunity.
Galectins (Gal) are a family of proteins characterized by a ~130 amino acid carbohydrate-recognition domain (CRD) that binds β-galactosides1-4. Their ligands are found widely and at high density in the glycoprotein products of the secretory pathway5,6. The galectin gene family expanded early with the radiation of multicellular eukaryotes7, along with the genes encoding Golgi N-acetylglucosaminyltransferases and β-galactosyltansferases that generate the galectin binding site on the Asn (N)-linked glycans of transmembrane glycoproteins.
Galectin are synthesized in the cytosol and also localized to the nucleus and cell surface. A primordial function of galectins may have been the capacity to cross-link β-galactoside-containing glycoconjugates on pathogens, to engulf and destroy them8. In mammalian cells, some galectins in the cytosol act as a danger receptor that restricts the proliferation of invasive bacteria. For example, Salmonella enters eukaryotic cells and disrupts lysosomal vesicles which exposing the luminal N-glycans of lysosomal-associated membrane glycoprotein (LAMP) to the cytosol. The N-glycans recruit galectin-8 (Gal-8) and the NDP52 complex leading to polyubiquitination and autophagy of damaged Salmonella-containing vacuoles9-11. The N-terminal CRD of Gal-8 binds β-galactosides, and the C-terminal CRD also binds a nine amino acid sequence in NDP52, thus recognizing both the peptide and β-galactosides. Although, specificity of galectins for β-galactosides has been largely conserved, CRD affinities are relatively low which increases the chemical space for evolution of non-conventional ligands that mimic the β-galactoside. However, in many cases, evolution does not move towards higher affinity and specificity, but rather a function endpoint. Low CRD affinities for the ubiquitous β-galactosides and dynamic cross-linking of glycoproteins are precisely the biophysical features required for galectins to function in many setting as discussed below.
Galectins are classified into the prototype (Gal -1, -2, -5, -7, -10, -11, -13), tandem-repeat (Gal -4, -6, -8, -9, and -12), and chimera-type (Gal -3) groups. Although galectins with single or dual CRD can self-associate, Gal-3 differs from the others, with one CRD and a N-terminal proline- and glycine-rich domain ~120 amino acid12 which is intrinsically disordered (ID) and drives Gal-3 self-association, stimulated by increasing local concentrations of multivalent ligands13,14.
The inclusion of the ID domain is required for phase transition when Gal-3 is mixed with bivalent N-glycan ligand at optimal concentrations in vitro. Self-association increases the capacity for Gal-3 to cross-link with transmembrane glycoproteins, which often have multiple N-glycosylation sites and act as multivalent partners in lattice formation13,15,16. The lattice is generated by multiple and simultaneous interactions of multi-/bi-valent galectins with diverse cell surface glycoproteins. Single CRD galectins may also dimerize as exemplified by Gal-117.
At higher concentrations, cross-linking results in a reversible phase transition with properties as described for liquid droplets (e.g. stress granules)18,19. In addition to multivalency and intrinsic disorder, the galectin lattice liquid-droplet properties include stochastic binding with rapid exchange, viscosity and surface tension, a complex composition of interactors, and affinity regulation by Golgi N-glycan remodeling20.
In live cells, the galactin lattice is a planar liquid-droplet, held in proximity to the membrane by transmembrane glycoproteins. The diversity of glycoproteins and the flexiblity of glycan polymers contributes to a disordered geometry 21,22 (Fig. 1). The Gal-3 CRD displays an increase in conformational entropy upon binding to N-acetyllactoasamine disaccharide units (LacNAc or Galβ1-3/4GlcNAc) which is unusual in higher-affinity receptor ligand interactions. The transfer of binding entropy to the larger CRD domain may compensate for loss of entropy in the bound N-glycan, and thereby contributes favorably to the rapid on-off and exchange of ligands23,24. Gal-3 and Gal-9 have been shown to slow lateral diffusion of glycoprotein receptors measured by fluorescence recovery after photobleaching (FRAP)25. For example, changes in Gal-9 levels, N-glycan branching, or rates of endocytosis altered the FRAP half-life of glucagon receptor on primary liver cells in a manner consistent with protecting the receptors from loss to endocytosis26. Gal-3 oligomerization at the cell surface has also been measured by fluorescence resonance energy transfer (FRET)27, and Gal-3 lattice dynamics of β1 integrin receptor by single-particle tracking28.
In a reversal of roles, the galectin lattice can interpose into more stable glycoprotein complexes and increase entropy and thereby turnover. The addition of recombinant Gal-3 to tumor cells stimulates turnover of β1-integrin in focal adhesion, signaling and cell motility at an optimal concentration, above which Gal-3 is inhibitory29,30. Endogenous Gal-3 in mice is required for normal neutrophil motility into sites of fungal infection31. A bell-shaped response is also observed with increasing substratum stiffness; an additional factor that interacts with the galectin lattice to regulate cell motility32. Optimal proportions of interactors and reversibility are request features of phase-transition systems.
Among other variables, the density and stiffness of the extracellular matrix interacts with cellular N-glycan remodeling to determine focal adhesion turnover rates32. Similar nonlinear effects on cell motility have been observed with recombinant Gal-833,34. Gal-3 enhances the dynamics at the immune synapse, another microfilament-anchored cell-cell connection between T cells and antigen-presenting cells. Gal-3 interactions with glycosylated receptors in the synapse provides exploratory delay and threshold for TCR contacts to trigger T cells activation35-37.
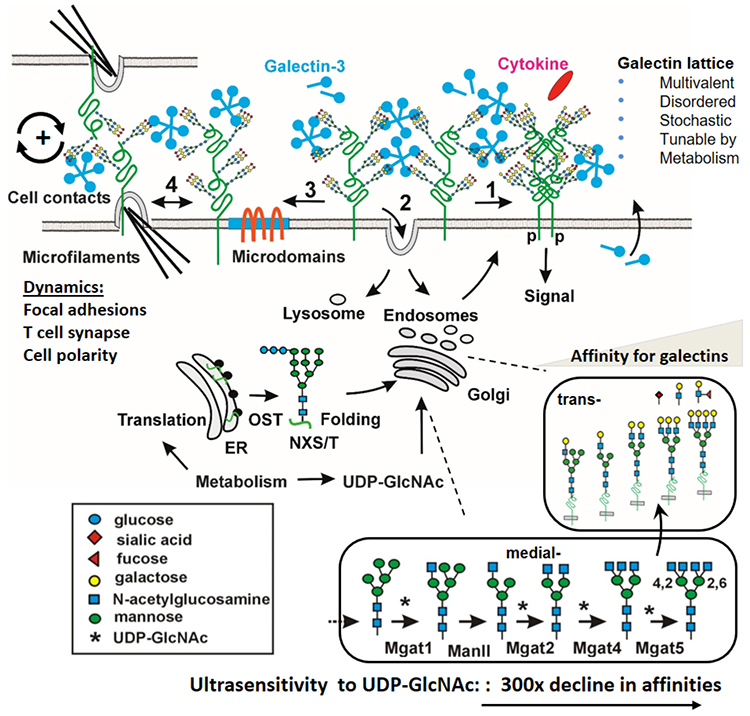
The affinities of CRDs for the LacNAc disaccharide [Galβ1-3/4GlcNAc] are in the 10-100 μM range38, but binding avidity is greatly increased with LacNAc density39,40. Mutations in Golgi enzymes that selectively eliminate LacNAc in all N-glycans reduce Gal-1, -3 and -8 binding to cultured mammalian cells by ~90%, indicating that N-glycans are the major Gal ligands in these cells41.
N-glycosylation sites [NXS/T(X≠P)] are genetically encoded in the protein sequence, and differ between glycoproteins. The number and position of sites on the protein fold influences the interaction with Golgi remodelling enzymes42-44, and thereby the potential for cross-linking by galectins. However, most glycosylated sites are occupied with a biosynthetically-related distribution of N-glycan structures, beginning with Golgi N-glycan branching, poly-LacNAc extension and capping with fucose and sialic acid. LacNAc density depends on the number of N-glycan sites, expression of Golgi N-acetylglucosaminyltransferases (encoded by MGAT1, MGAT2, MGAT4a,b,c, MGAT5), and the supply of nucleotide-sugar substrates45 (Fig. 1).
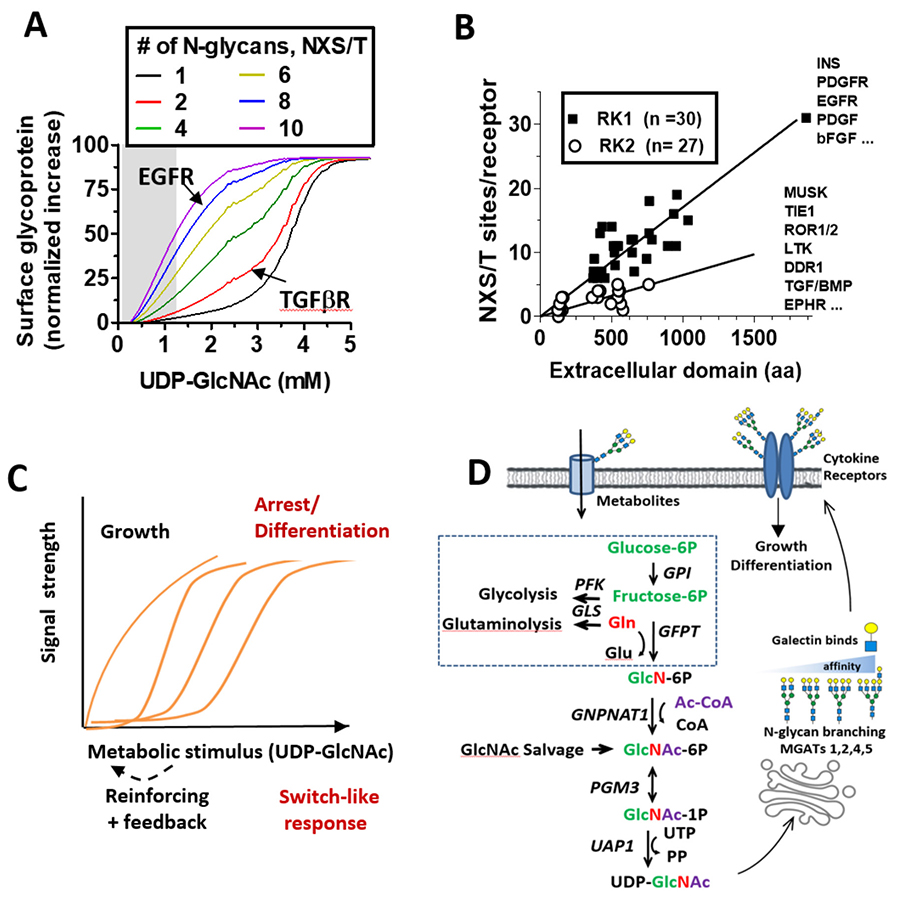
Developmental programs of gene expression and environmental conditions determine the cumulative output of the Golgi pathway46. However, heterogeneity is commonly observed at each site, and is due to competition between enzymes for acceptor substrates47, nucleotide-sugar levels48, as well as cooperative association between enzymes49, and variation in transit rates of glycoprotein substrates in the Golgi50. Glycoproteins with n modification NXS/T(X≠P) sites and X possible structures can generate an exponential number (Xn) of possible isoforms. For example, the epidermal growth factor receptor (EGFR) has eleven N-glycosylation sites and eight are modified and remodeled in the Golgi51. Using a conservatively estimate of only 15 possible N-glycan structures at each site, results in 158 = 2,562,890,625 possible glycoforms; too many to evaluate individually52. This number is more than three orders of magnitude higher than the number of EGFRs displayed on a cell surface at a given time (~1-5x105)53.
However, the problem is substantially simplified if functionally equivalent glycans can be binned by similar affinities for Gal-3. This reduces the X component of the above formula and provides a means of computing galectin-glycoprotein interactions45. More specifically, galectin binding and associated phase-transition at the cell surface largely depends on LacNAc content/density rather than the presence or absence of single glycoforms. In this manner, defining LacNAc content within glycoforms, rather than cataloguing distinct structures, is the much more relevant approach to deciphering the functions of galectins, and perhaps other multistep post-translational modifications21. With stress- or developmental- induced changes in these parameters, the effects on glycoform distribution and interactions with galectins can be calculated and used to predict changes in receptor signaling and cellular behavior45.
A critical regulator of LacNAc content, and in turn galectin affinities for glycoproteins, is metabolic supply of UDP-GlcNAc by the hexosamine biosynthetic pathway (HBP) to the Golgi N-glycan branching pathway45,54,55 (Fig. 2D). In silico simulations and experimental studies confirmed that the N-glycan branching pathway is ultrasensitive (Hill coefficient, Hn) to UDP-GlcNAc, with a ~300 fold decline in affinity for UDP-GlcNAc from MGAT1 to MGAT545. The tri- and tetra- antennary products increases in sigmodal or switch-like manner as UDP-GlcNAc concentrations increase in the medial Golgi (Fig. 2C). The GlcNAc antennae are substrates for β1,4galactosyltransferases, which generate LacNAc antennae that can be extended in the trans Golgi to poly-LacNAc of varying lengths. Importantly, the model predicts that a gain of MGAT1 activity reduces the availability of UDP-GlcNAc to MGAT4 and MGAT5 enzymes, thus reducing the synthesis of higher-affinity Gal-3 ligands (tri- and tetra-antennary). This counter intuitive feature was confirmed in cancer and non-transformed cells, in T cell activation in vitro, and with gain-of-function MGAT1 human polymorphisms and biochemical analysis in autoimmune disease45,56.
Co-evolution of the branching pathway ultrasensitivity and its glycoprotein substrates provides us with additional clues to galectin lattice functions. Receptors with low site number are recruited at a later stage in the titration of UDP-GlcNAc and branching relative to receptors with high site multiplicity45. Site number and density in cytokine receptors has evolved to promote growth signaling followed by arrest signaling as a function of UDP-GlcNAc levels. For example, EGFR with eight occupied sites, stimulates metabolism including UDP-GlcNAc biosynthesis, and this ultimately recruits TGF-β receptors with only two N-glycans into the lattice allowing increased autocrine TGF-β/Smad2/3 signaling and suppresses proliferation in non-transformed cells45. The glucose transporter GLUT4 has only one N-glycan and titration of intracellular UDP-GlcNAc increased GLUT4 cell surface expression with a switch-like response matching the global increase in tetra-antennary N-glycans (Fig. 2A, black line)45,56.
Insulin release by pancreatic β-cells is also dependent on N-glycan branching on GLUT2 transporters and the galectin lattice57,58. GLUTs 1 to 11 have a single glycosylation site, consistent with a requirement for switch-like engagement with the galectin lattice and regulation by UDP-GlcNAc production via the HBP. Elevated expression of Mgat4, Mgat5 and HBP activities are often observed in cancer cells59,60, which suppresses both pathway ultrasensitivity to UDP-GlcNAc and negative regulation of growth61. Thus, systems where (NXS/T(X≠P) number differs substantially between receptor groups that signal in opposite directions creates a UDP-GlcNAc-dependent delay between their recruitment to the lattice (Fig. 2B). Examples include the hepatic glucagon receptor and glucose homeostasis26,62.
Thus, N-glycan biosynthesis and biophysical properties of the galectin lattice provide a selective advantage to animals in the form of tunable and either graded or switch-like stimulus-response relationships (Fig. 2C)21. Our phylogenetic analysis of N-glycosylation sites has served as a model for how post-translational modification, more broadly, can evolve rapidly under selection and provide insight on function 63.
In T cells, the galectin lattice regulates the threshold for T cell receptor (TCR) clustering and signaling at the immune synapse19. Optimal galectin lattice dynamics in the thymus is required to generate T cells via positive selection by controlling CD4/CD8 co-receptor surface retention and TCR clustering/signaling19,37,64. Differentiation of peripheral T cells into pro-inflammatory T helper 1 and T helper 17 over anti-inflammatory regulatory T cell responses is inhibited by the galectin lattice, which combined with TCR regulation determines risk of autoimmune disease19,54,56,64-67. Similar regulation has been observed in B cells, where minimal expression of branching is required to maintain the CD19 co-receptor at the cell surface and allow B cell receptor responses, while increasing lattice strength suppresses pro-inflammatory innate Toll-like receptor responses to control B cell-dependent autoimmunity68,69.
In humans, single nucleotide polymorphisms (SNPs) in MGAT1 and MGAT5 are associated with Multiple Sclerosis (MS)56,70. Gain-of-activity SNPs in the coding sequence of MGAT1 increase expression and when coupled with a ~300 fold higher affinity for UDPGlcNAc, this results in elevated MGAT1 expression depriving MGAT5 of substrate to weaken LacNAc-galectin dynamics and promote pro-inflammatory T cell and B cell responses. Vitamin D3 deficiency, a well-known risk factor for MS and other autoimmune diseases, alters MGAT1 mRNA levels to lower branching and cause a hyper active T cells phenotype. Vitamin D3 supplementation has the opposite effect, strengthening the galectin lattice to suppresses autoimmunity56,71.
The gain-of-activity MGAT1 SNPs and a CTLA-4 SNP that reduces N-glycan site occupancy at one of two sites, additively increase the risk of MS. Due to pathway ultrasensitivity, gain-of-activity MGAT1 decreases UDP-GlcNAc availability to MGAT5 and together with the loss of a glycosylation site, CTLA-4 is only weakly recruited to galectin lattice at the cell surface, thereby delaying its action as a negative regulator of T cell proliferation45. CTLA4 receptor binds CD80 and CD86 in the immune synapse and dampens TCR co-receptor CD28 activity. CTLA-4 levels are rescued by GlcNAc supplementation in tissue culture, and in vivo oral GlcNAc also suppresses experimental autoimmune disease in mice and rescues signaling consistent with the model65,66.
LacNAc epitopes for galectin binding occur at various positions in structurally distinct N-glycans, and experimental evidence suggests that losses can be compensated by gains at another position in the same glycan or between glycans on glycoproteins with multiple sites. Bio-equivalence of binding sites occurs within and between glycans on a protein with multiple sites of glycosylation18,26,45,54. For example, a tetraantennary glycan has a similar affinity for Gal-3 as triantennary with one or two poly-LacNAc units38. At the level of glycoforms, two sites each with triantennary glycans is similar to a bi- and a tetra- antennary45. As an example at the cellular level, Mgat5 has no gene homologues, but supplementing cultured Mgat5 deficient cells with GlcNAc completely rescues surface EGFR in association with Gal-3 and by increasing UDP-GlcNAc levels and thereby the activities of Mgat4 and the β3GNT family of poly N-acetyllactosamine extension enzymes45,72. GlcNAc is taken up by fluid phase endocytosis and salvaged into HBP increasing UDP-GlcNAc levels and compensating LacNAc content in N-glycans45.
Bio-equivalence is also an important consideration in T cells and autoimmunity72. MGAT2 deficient T cells display marked increases in poly-LacNAc extension of the single GlcNAc branch in hybrid N-glycans catalyzed by trans- Golgi β3GNT enzymes, which maintains LacNAc content and thereby compensates for loss of bi- and tri-antennary structures. Remarkably, increased poly-LacNAc extension is due to the flux of unused UDP-GlcNAc from the medial to trans Golgi, as the MGAT2 mutation blocks branching beyond MGAT1 (Fig. 3). The increase in trans Golgi UDP-GlcNAc levels enhances β3GNT enzyme activity without altering its gene expression. The evolved compartmentalization of branching enzymes in the medial Golgi and β3GNT extension enzymes in the trans-Golgi provides a fail-safe mechanism to maintain LacNAc content within N-glycans72. This bio-equivalence mechanism plays a critical role in regulating the sensitivity of T cells to autoantigens and prevention of autoimmunity in mice.
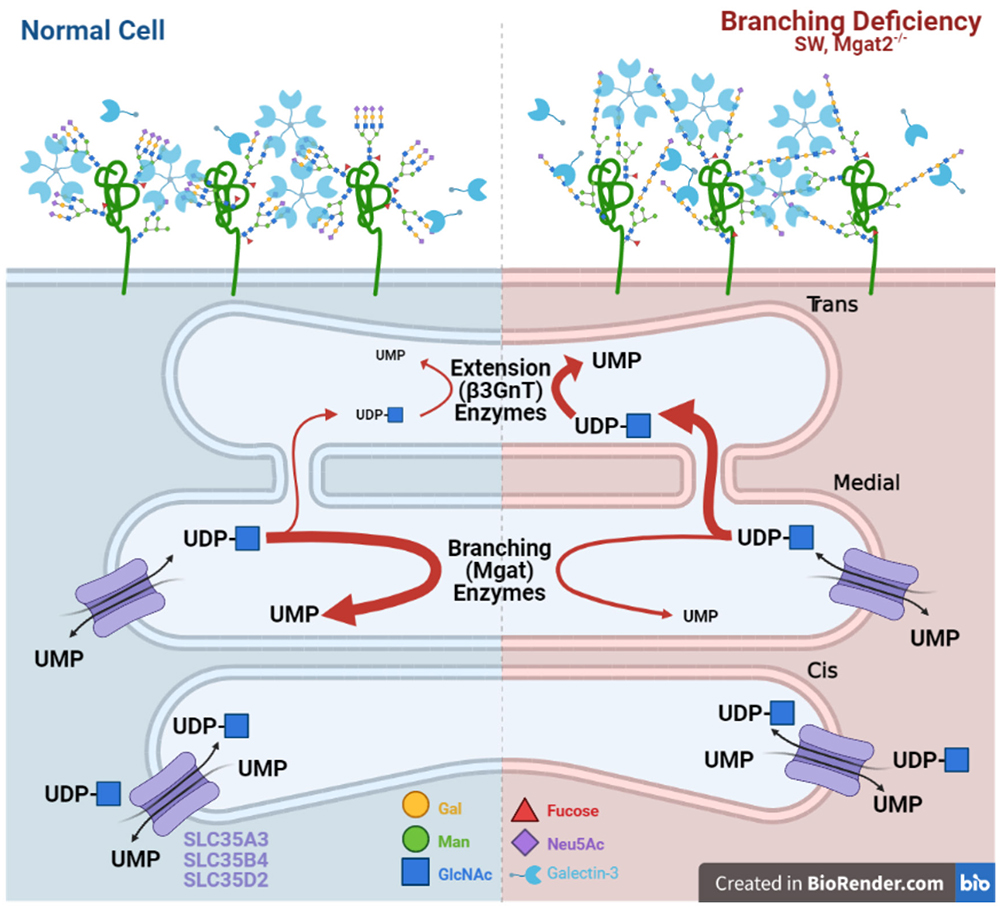
Nevertheless, these experiments also raise questions about the nature of bioequivalence. In particular, whether bio-equivalence can be understood at the global level and through the study of glycan-lectin interactions or whether it must be investigated for each individual glycoprotein. The poly-LacNAc compensation induced by MGAT2 deficiency does not uniformly rescue biological function. Germline deficiency of MGAT2 results in a significantly more severe phenotype than Mgat5 disruption73,74. Furthermore, although T cell activation thresholds in peripheral T cells are essentially identical between MGAT5 and MGAT2 deficient T cells, T cell development with MGAT2 deficiency demonstrates a significantly more severe phenotype than in MGAT5 deficient mice37,56.
These discrepancies can be explained by a number of different scenarios. Poly-LacNAc compensation of a particular length may indeed be bioequivalent to tri-antennary N-glycans but may only occur on some receptors or in some cell types and not others. Indeed, in MGAT2 deficient mice, binding of the poly-LacNAc specific Lycopersicon esculentum lectin/agglutinin (LEL/LEA) to peripheral T cells and double positive thymocytes was similar despite ~10-fold higher N-glycan branching in the latter37,56. This suggests that the degree of poly-LacNAc extension in MGAT2 deficient mice may only efficiently compensate in cells with low branching activity such as peripheral T cells. Cell types that rely on higher branching levels may not produce adequate extension to compensate for the loss of branching and would produce the T cell development differences observed between MGAT5 and MGAT2 deficient mice.
Alternatively, poly-LacNAc biosynthesis may be globally adequate, but only result in bio-equivalence for some receptors and not others. This would suggest poly-LacNAc addition is substrate dependent at the level of the glycoprotein and its individual sites and therefore bio-equivalence may not be generalized across different glycoproteins. Importantly, in future studies, these two scenarios may be broadly distinguished by performing galectin binding experiments on different cell types from these mice.
As first described for cancer by Otto Warburg nearly a century ago, rapidly proliferating cells, including activated T cells, switch from oxidative phosphorylation to aerobic glycolysis plus glutaminolysis. This markedly increases glucose and glutamine consumption by anabolic pathways in the cells. Both glucose, and glutamine as an amine donor, are required by the HBP for de novo synthesis of UDP-GlcNAc and therefore are limiting for N-glycan branching in non-transformed cells under normal conditions45,54. Thus, increasing aerobic glycolysis and glutaminolysis inhibits the flux of glucose and glutamine to UDP-GlcNAc biosynthesis and thereby reducing galectin lattice strength and receptor content (Fig. 2D).
In antigen-stimulated T cells, aerobic glycolysis and glutaminolysis cooperatively reduce de novo UDP-GlcNAc, branching and lattice strength to drive T cell proliferation and pro-inflammatory TH17 over anti-inflammatory T regulatory (iTreg) differentiation by lowering IL-2Rα (CD25) surface levels54,66. In this manner, aerobic glycolysis and glutaminolysis serve as critical negative regulators of galectin lattice strength, IL-2Rα receptor at the cell surface, and cell fate. However, GlcNAc is salvaged into the HBP to generate UDP-GlcNAc when provided in tissue culture and orally in mice, circumventing these rate-limiting steps in the HBP and driving therapeutically targeted changes for autoimmune diseases like MS. Programmed catabolism of glycoconjugates during development and disease may increase GlcNAc availability, thereby galectin lattice strength and receptors controlling the levels of TH17 and iTreg cells. For example, the severity of lung impairment with influenza infection correlates with early accumulation of hyaluronan, and treatment with hyaluronidase, which releases GlcNAc, results in faster recovery of body weight and lung function75. Further work is required to explore the turnover of glyconjugates, and the role GlcNAc salvage.
The galectin lattice drives myelination by promoting surface retention of platelet-derived growth factor receptor (PDGFR) in oligodendrocyte precursor cells, a receptor that plays a critical role in oligodendrogenesis76. Disrupting the lattice via whole animal deletion of Gal-3 or inducible knockout of the Mgat1 branching enzyme only in oligodendrocyte precursor cells leads to decreased production of oligodendrocytes, and reduced primary myelination of axons in mice76,77.
Similarly, loss-of-function mutations of phosphoacetylglucosamine mutase (PGM3) in humans, a key enzyme in the HBP pathway for production of UDP-GlcNAc, display reduced branching and severe hypomyelination78. Moreover, feeding GlcNAc to lactating female mice drove primary myelination in the nursing pups after being secreted in breast milk, systemically absorbed in the pups’ bowel, crossing the blood brain barrier and being metabolized to UDP-GlcNAc in the brain76. Interestingly, GlcNAc is a major component of human breast milk oligosaccharides79 that can be released as a monosaccharide by infant microbiota80. In contrast, GlcNAc is not a significant component of commercial baby formula, raising the possibility that the known association of increased myelination and cognitive function with breast-feeding may in part be due to GlcNAc in human breast-milk.
In MS patients, failure of re-myelination following demyelination is common and leads to ongoing axonal damage, neurodegenration and progressive disability. In a mouse model of MS, providing mice with oral GlcNAc following demyelination drove myelin repair, reduced axonal damage, and enhanced functional recovery. In humans, MS is characterized by two primary subtypes, namely episodes of inflammatory demyelination in ‘Relapsing-remitting MS’ and slow progressive neurodegeneration in ‘Progressive MS’. Remarkably, serum levels of N-acetylhexosamines (HexNAc), which includes GlcNAc and its stereoisomers GalNAc and ManNAc, are markedly reduced in progressive MS patients compared to healthy controls and patients with relapsing-remitting MS81. Moreover, lower HexNAc serum levels correlated with multiple measures of neurodegeneration in MS, namely worse expanded disability status scale scores, lower thalamic volume, and thinner retinal nerve fiber layer. Low baseline serum levels also correlated with a greater percentage of brain volume loss at 18 months.
Thus, coupled with its negative regulation of pro-inflammatory T and B cell responses, oral GlcNAc is uniquely positioned to reverse four major mechanisms driving MS pathogenesis and severity, namely pro-inflammatory T-cell responses, pro-inflammatory innate B-cell activity, failure of myelin repair, and neurodegeneration. Such diverse mechanisms of action are not present in current MS therapies, which target inflammatory responses but not myelin repair or neurodegeneration. This suggests that GlcNAc may be therapeutically beneficial to MS patients. To pursue this possibility, oral GlcNAc in MS patients is currently in an open-label clinical trial led by Dr. Michael Demetriou. Oral GlcNAc was well tolerated, raised serum GlcNAc levels and may have induced clinical benefit. Additional blinded trials are planned to confirm these findings.
The galectin lattice model is a powerful and predictive framework with which to approach complex glycobiology. It provides insight into both the role of glycosylation in cell surface dynamics as well as the regulatory logic of glycoprotein biosynthetic pathways. However, many questions regarding its structure, function, regulation and dysregulation remain open. For instance, better methods of high-resolution mapping of the weak interactions inherent in galectin-glycoprotein interactions is required to study the contribution of galectin family members and possible subnetworks within the galectin lattice. Proteomic approaches fail to capture the native cell surface lattice, while low throughput approaches focusing on individual receptors do not address the larger network of interaction between signaling pathways. As resolution improves from the glycoprotein to the glycoforms distribution, new and exciting insights of the galectin lattice are likely to emerge and contribute to our understanding of development, aging and disease.
J.W.D.’s research is supported by grants from the Canadian Institutes of Health Research (MOP-126183, MOP-136789, MOP-126029), and Canada Research Chairs Program. M.D.’s research is supported by the National Institute of Allergy and Infectious Diseases (R01 AI108917) and the National Center for Complementary and Integrative Health (R01 AT007452).



