
Shinnosuke Wakamori
Ph.D.
Assistant Professor, Laboratory of Molecular Architecture, Department of Chemistry for Life Sciences and Agriculture, Faculty of Life Sciences, Tokyo University of Agriculture.
Assistant Professor, Kwansei Gakuin University (Prof. Hidetoshi Yamada and Associate Prof. Kei Murakami), 2018–2021; Postdoctoral fellow, Kwansei Gakuin University (Prof. Hidetoshi Yamada), 2016–2018; Researcher, Shin-Etsu Chemical Co., Ltd., 2014–2016; Ph.D., The University of Tokyo (Prof. Hidenori Watanabe), 2014; B.S., Waseda University (Prof. Nobuhiro Kanomata), 2009.
Cyclodextrins (CDs) are cyclic oligomers of α-1,4-D-glucopyranoside. Because the central cavities of CDs can be used to encapsulate small molecules, cyclic hexamer to octamer (CD6–8) have been widely used. While CD-hundreds are known for large CDs, the synthesis of the only CD5 had been only one reported for small CDs. Smaller CDs, such as CD4 and CD3, have never been synthesized because their molecular sizes are too small for the pyranose ring to adopt a stable chair-type conformation. In this report, we describe the chemical synthesis of both CD4 and CD3. The development of a specific bridging group between the 3- and 6-oxygen positions (O-3 and O-6) of D-glucose led to the successful synthesis of these compounds. In other words, the adoption of this bridging group provides both the stereoselective glycosylation reaction and the supple conformation of the pyranose ring, which are required for the synthesis of CD4 and CD3.
Cyclodextrins (CDs) are cyclic oligosaccharides composed of α-1,4-linked D-glucose.1 In particular, cyclic hexamer to octamer (CD6–CD8), which are easy to prepare and are inexpensive via bulk production using enzymes, are mainly known (Fig. 1).2 Because these compounds are only composed only of non-toxic D-glucose3–5 and their hydrophobic central cavities can capture small molecules, and hence, they are widely used in industrial products, pharmaceuticals, and also consumer products.
Since the type of compound that can be encapsulated depends on the number of D-glucose components in CDs, many studies have focused on the size of CDs. The structures of CDs larger than CD8 have been confirmed up to CD35,6 while the chemical synthesis of CD5 is the smallest of the CDs reported to date.7 Smaller CDs, such as CD4 and CD3, have being theoretically studied without confirming their existence. In 1957, French reported that a space-filling model of a CD with an infinite number of glucose molecules could be constructed from CD3, but that the smallest CD obtained upon treating glycogen with Bacillus macerans, a type of eubacterium, was CD6.8 In 1970, Sundararajan and Rao stated on the basis of chemical calculations that CDs smaller than CD6 can not be cyclized.9 Nakagawa and co-workers achieved the chemical synthesis of CD5 in 1994 7 and in the following year, Immel and co-workers estimated that the synthesis of CD4 and CD3 would be extremely difficult due to the inability of the pyranose ring in the constituent D-glucose molecules to form the most stable chair conformation (4C1 conformation) (Fig. 1)10. Despite the synthesis of several CD-like molecules with different binding types,11–14 the synthesis of CD3 and CD4, which are composed of only α-1,4 bonds with strained D-glucose, remains an unmet goal.
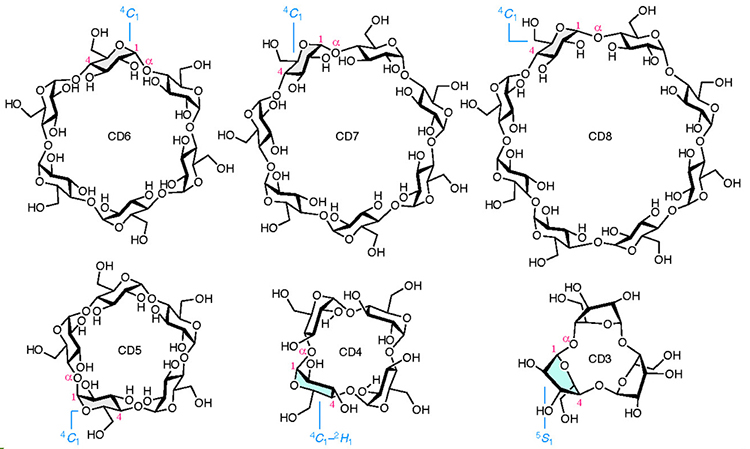
Herein, we succeessfully carried out the chemical synthesis of CD4 and CD3, thereby, providing their existence.15 The main feature of this study is the specific bridging group introduced into the 3- and 6-oxygen positions (O-3 and O-6) of the D-glucose derivatives. In this paper, the pyranose ring whose conformation was not determined is displayed as a planar structure, and the ring whose conformation was determined using the method described below is drawn in a steric structure.
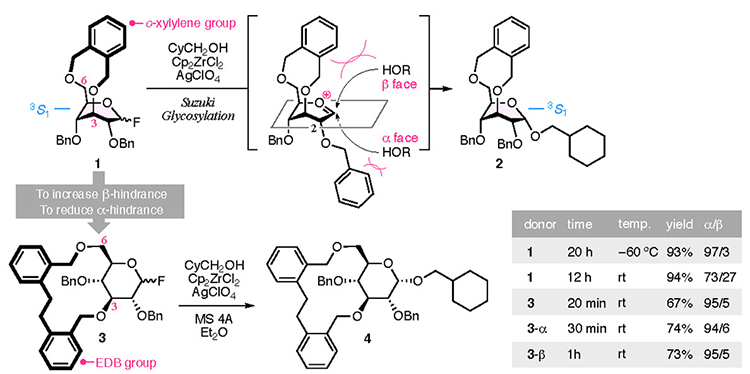
The presence of a bridging group enables the highly α-selective glycosylation reaction required for the CD synthesis to occur (Fig. 2). Our previous study has reported an o-xylylene bridging group,16,17 Suzuki glycosylation18 using zirconocene dichloride and silver perchlorate, allowed the kinetic reaction of o-xylylene bridged 1 and cyclohexanelmethanol to occur, with high α-selectivity. However, the bridge locks the pyranose ring into the 3S1 conformation, which directs the 2-O-benzyl group axially toward the α face, thus inducing adverse steric hindrance. The α-selectivity of glucosides 2 therefore decreases with an increase in the reaction temperature. This issue was resolved by increasing the steric hindrance at the β face and reducing the overhang of the 2-O-benzyl group on the α face. To satisfy both of these requirements, we developed the 1,1′-(ethan-1,2-diyl)dibenzen-2,2’-bis(methylene) (EDB) group as a bridging group, which has higher flexibility than the o-xylylene group. The glycosylation reaction using EDB-bridged glucosyl fluoride 3 afforded glucoside 4 with good α-selectivity even at room temperature within 20 min. The reaction proceeded through an corresponding oxocarbenium ion intermediate and similar α-selectivities were observed for 3-α and 3-β.
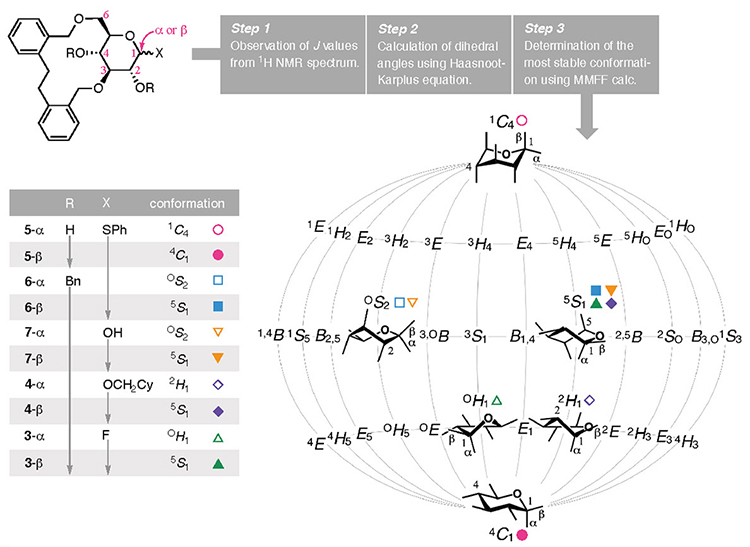
Another feature of bridging group developed in this study is that the pyranose moiety can adopt various conformations. The attachment of a bridging group on the pyranose ring produces a bicyclic skeleton in which the newly formed ring modulates the conformation of the pyranose scaffold. As a result, the conformation of the pyranose ring in D-glucose was converted from its most stable chair-type conformation (4C1) to various conformations. A short bridge "locks" the con- formation into a motif with more axial substituents, as seen in 1 and 219 and others.20–24 In this study, the pyranose conformation was modified when employing the EDB group as the bridging group due to the subtle alteration in its structure, as revealed by the 1H NMR coupling constants observed for 3 to 7 (Fig. 3). The following method was developed to determine the steric configurations of the pyranose rings bridged the EDB group. The coupling constants were obtained from the 1H NMR spectrum (Step 1), and the dihedral angles were calculated using the Haasnoot–Karplus equation (Step 2).25 Because one coupling constant gives up to four solutions when using this equation, the most stable conformation among the available combinations of dihedral angles was determined from using Merck molecular force field (MMFF) calculations (Step 3). By analyzing the obtained conformations, 5-α (R = H and X = α-SPh) adopted a 1C4 comformation, whereas its anomeric isomer, 5-β, possessed the same chair type but with a 4C1 conformation. For 6-α and 6-β, where the two hydroxy groups were protected using benzyl groups (R = Bn), the OS2 and 5S1 conformations were assigned as a twist boat type. As displayed on a map of conformations that puts the 1C4 and 4C1 conformations on both poles (Cremer–Pople–Stoddart coordinates),26,27 the calculated conformations of the pyranose system in 3–7 were widely distributed, as shown in Fig. 3. We propose that this phenomenon is the result of the moderate constraint imposed by the EDB group competing with the pyranose ring's tendency to adopt a stable chair-type conformation. Thus, the EDB bridging group makes the pyranose ring "supple" and, in combination with the α-selective glycosylation reaction using glucosyl fluoride 3, enables the synthesis of CD4 and CD3.
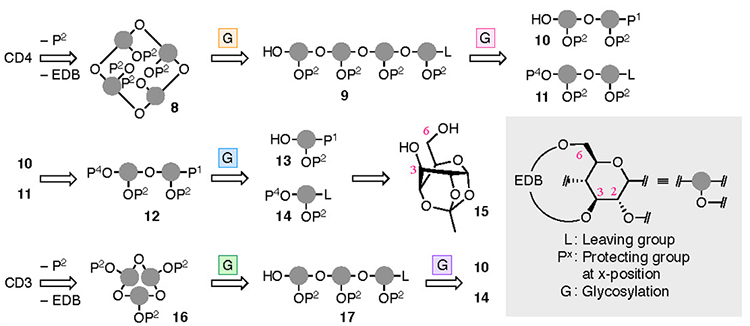
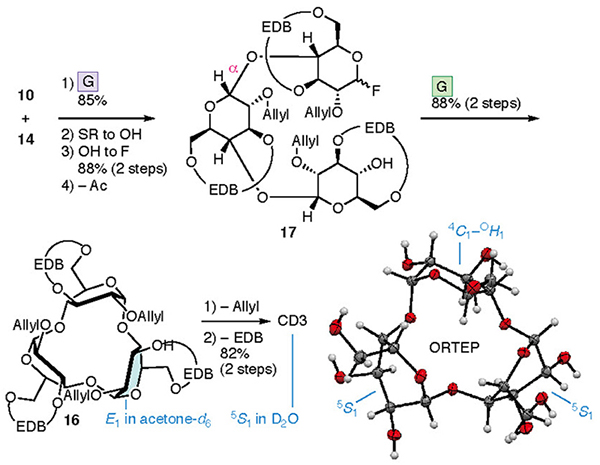
The synthesis of CD4 and CD3 is shown in Fig. 4. In the chemical synthesis of CD4 and CD3, which consist of repeating α-1,4 bonds of D-glucose, the oxygen atoms other than those in the α-1,4 bonds were protected. Since the EDB group already protects O-3 and O-6, the protection of O-2 (P2) was carried out. CD4 was obtained upon full deprotection of a cyclized intermediate 8, which was obtained via intramolecular glycosylation of a linear tetramer 9. Tetramer 9 was produced upon the glycosylation of dimers 10 and 11, manipulation of the protecting groups, and the addition of a leaving group. Dimers 10 and 11 were derived from monomers 13 and 14, respectively. To introduce the EDB bridges in 13 and 14, which the synthesis began with 1,2,4-orthoacetylglucose (15),28 in which the O-3 and O-6 are locked in the same direction so as to ease the construction of the bridge. During the synthesis of CD3, precursor 16 was produced via an intramolecular glycosylation of a linear trimer 17 which was formed from the dimer 10 and the monomer 14. Using a fluorine atom as a leaving group (L) allowes the α-selective glycosylation reaction required for synthesizing the CDs to occur.29,30 The suppleness caused by the EDB bridge enables the cyclization to occur, leading to 8 and 16, which may be impossible when the pyranose moieties adopt a 4C1 conformation.
The preparation of the disaccharide 12 is shown in Fig. 5. Etherification of 15 with 2,2′-bis-(bromomethyl)-dibenzyl31 furnished EDB-bridged 18. Maintaining the concentration of 15 at <10 mM ensured the reaction was reproducible. An indium bromide-promoted β-specific introduction of an arylthio groups, was accompanied by the cleavage of the orthoester 18. Subsequent removal of the acetyl group at O-2 provided 19 and 5-β, which were converted into 13 and 14, respectively. Glycosylation using 13 and 14 provided the dimer 12 with exellent α-selectivity. Higher selectivity was observed using 14 when compared to 3. Changing P2 from the benzyl group to a smaller allyl group reduces the steric hindrance at the α face by the alteration of P2. Transformation of 12 into 20, whose pyranoses adopt a 4C1 conformation, was confirmed by the coupling constant observed in the 1H NMR spectrum, which confirmed the α stereochemistry in 20, as shown in Fig. 5.

Cyclization of the linear tetramer gave CD4 (Fig. 6). Tetramer 21 was obtained from 10 and 11, both of which were derived from disaccharide 12. The stereochemistry of the newly formed glycosidic bond in tetramer 21 was undetermined because of the overlapping 1H NMR signals. Subsequently, fluorination of the anomeric position and removal of the acetyl group at O-4, followed by an intramolecular glycosylation reaction gave cyclic tetramer 8. The 1H and 13C NMR spectra of cyclized 8 were in agreement with the pattern of a monosaccharide, indicating the unified stereochemistry of the four anomeric positions. Since α-stereochemistry at the 1’ position was confirmed, anomeric linkages were all α. The reproducibility and yield of the intramolecular glycosylation were poor (six successes to 17 failures, <25% yield). Removal of the allyl group32 from the cyclic tetramer 8, followed by hydrogenolysis allowed the successful removal of the EDB group, which achieved the synthesis of CD4. The 1H NMR spectra revealed that the pyranose ring conformations were determined to be between 4C1 and 2H1 for CD4 and 2H1 for 8. The pyranose moiety in these compounds was distorted and flatter than observed in larger CDs.33–35
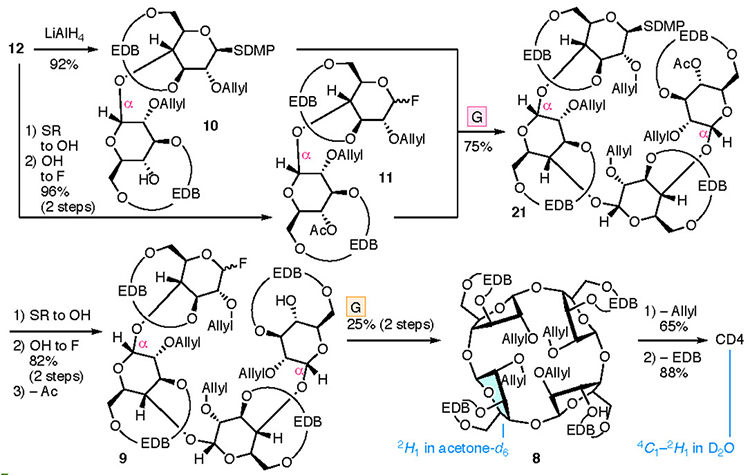
A similar process to that used to prepare CD4 led to the synthesis of CD3 (Fig. 7). Cyclization of the linear trimer 17 derived from dimer 10 and monomer 14 provided cyclic trimer 16. Intramolecular glycosylation was more efficient than that with 8, with the yield of product 16 reaching 88%. Removal of the allyl and EDB groups led to the formation of CD3. 1H and 13C NMR, and X-ray diffraction studies of a single crystal confirmed the structure of CD3. The NMR spectra of 16 and CD3 were consistent with equivalent sugar units and the average pyranose conformations were determined to be E1 and 5S1, respectively. The lower field shift of H-5 in the 1H NMR spectrum of CD3 may be a result of the deshielding effect induced by O-5. The X-ray diffraction analysis of a single crystal of CD3 indicated that the conformations of the three pyranoses were different (two with 5S1 and one between 4C1 and OH1 conformations) in the crystal lattice.
The use of glucose monomers with conformational flexibility has allowed for the synthesis of the strained CD4 and CD3. The creation of "suppleness" in sugars using a bridging group is potentially applicable to the synthesis of other strained compounds or situations where the molecule function requires a flexible structure. The averaged C3 and C4 symmetry observed in the NMR spectra of CD4 and CD3, where multiple stereocenters exist, can be useful towards the construction of molecular catalysts or metal-organic frameworks. CDs have general relevance for applications that take advantage of their capacity to hold molecules within their central cavity. We are currently investigating a method to supply CD4 and CD3 on a glam scale aiming for the further analysis of the physical properties and the development of the functions of CD4 and CD3.
This research was carried out in the Yamada Laboratory, Faculty of Science and Engineering, Kwansei Gakuin University. I thank Mr. Hirata, Mr. Ikuta, Mr. Kawasaki, Mr. Shimada, Mr. Hagimori, and Mr. Matsumoto, who have worked passionately on this research. I also thank Dr. Yusuke Tomabechi (School of Engineering, Tokai University) and Dr. Kazutada Ikeuchi (Faculty of Science, Hokkaido University) for their dedication and support.
Prof. Dr. Hidetoshi Yamada passed away suddenly on November 23, 2019. I express my regret on his untimely passing in the midst of his active career. I hope to preserve his unflagging respect for chemistry and shall always remember his sincere attitude towards research, and his passion towards everything that he did.



