 PDF版 |  この 作品 は クリエイティブ・コモンズ 表示 4.0 国際 ライセンスの下に提供されています。 |

氏名:西 望
1975年大阪大学理学部化学科卒業。1980年大阪大学大学院理学研究科後期課程(博士)終了。理学博士。
日本学術振興会奨励研究員、南カリフォルニア大学研究員、香川医科大学助手を経て香川大学総合生命科学研究センター准教授。この間、株式会社ガルファーマ社外取締役(2000年 — 2009年)。現在、香川大学客員研究員。
研究内容は、光合成細菌、前立腺/増殖因子、ガレクチンと変化しましたが、私の本質は古典的なタンパク質生化学者です。
このノートは、大腸菌を利用したヒトガレクチンファミリーの発現と精製に関する未発表データなどをまとめたものです。これまでガレクチンに馴染みがなく、新たに組換えガレクチンタンパク質の発現・精製を行う研究者(特に、生化学研究の経験が比較的浅い研究者)の参考になればと思います。
筆者が理研バイオリソースセンターに寄託・譲渡したガレクチンクローン(ヒト約60種類、マウス及びラット約20種類)を、Table 1に簡単な説明を付してまとめてあります。各クローンの情報には理研バイオリソースセンターのホームページからアクセスできます:理研バイオリソースセンター → 遺伝子材料開発室 > リソース情報 > 検索&リスト > 寄託者リスト → Nishi, Nozomu → 各クローン。詳しい説明が必要なクローンに関しては、対応するガレクチンの項に留意事項を記載しました。Table 1にはpTrc-HisB vector(ポリヒスチジン配列を含む分子量4,000 ~ 5,000のタグがN-末端側に付加される、大腸菌用発現ベクター)を使用したものが含まれていますが、ほとんどの場合、ガレクチンの発現にこれらのクローンを利用するメリットはありません。本文や図表中で使用するガレクチンの略称(例:ガレクチン−1、G1)については、Table 1の右端の欄を参照して下さい。Table 2は、組換えタンパク質の分子量などの情報をまとめたものです。このノートで使用するアミノ酸残基番号は、特に注意書きがない限り、開始コドンに対応するメチオニン残基を1としています。
プロトタイプ型ガレクチンは1個の糖鎖認識ドメイン(carbohydrate recognition domain, CRD)のみで構成されますが、キメラ型ガレクチン(G3)はN-末端側の非糖鎖認識ドメイン(コラーゲン様ドメイン)+ CRD、タンデムリピート型ガレクチン(G4, G8, G9, G12)はN-末端側CRD + リンカーペプチド + C-末端側CRDから成ります。G3のコラーゲン様ドメインとCRD、あるいはタンデムリピート型ガレクチンにおけるCRDとリンカーペプチドの境界を厳密に決定することは困難なので、以下に記載するCRDの領域もある程度の曖昧さを含むものです。また、ここで使用するCRDという名称は、糖鎖結合に必須な最小単位を意味するのではなく、プロトタイプ型ガレクチンであれば、その分子全体を指しています。
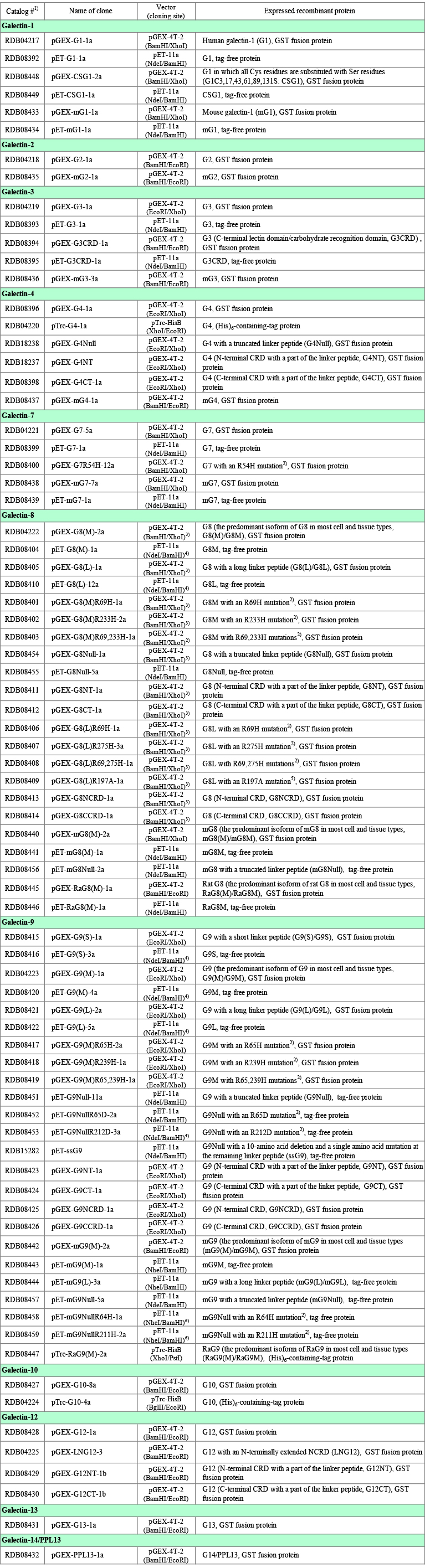
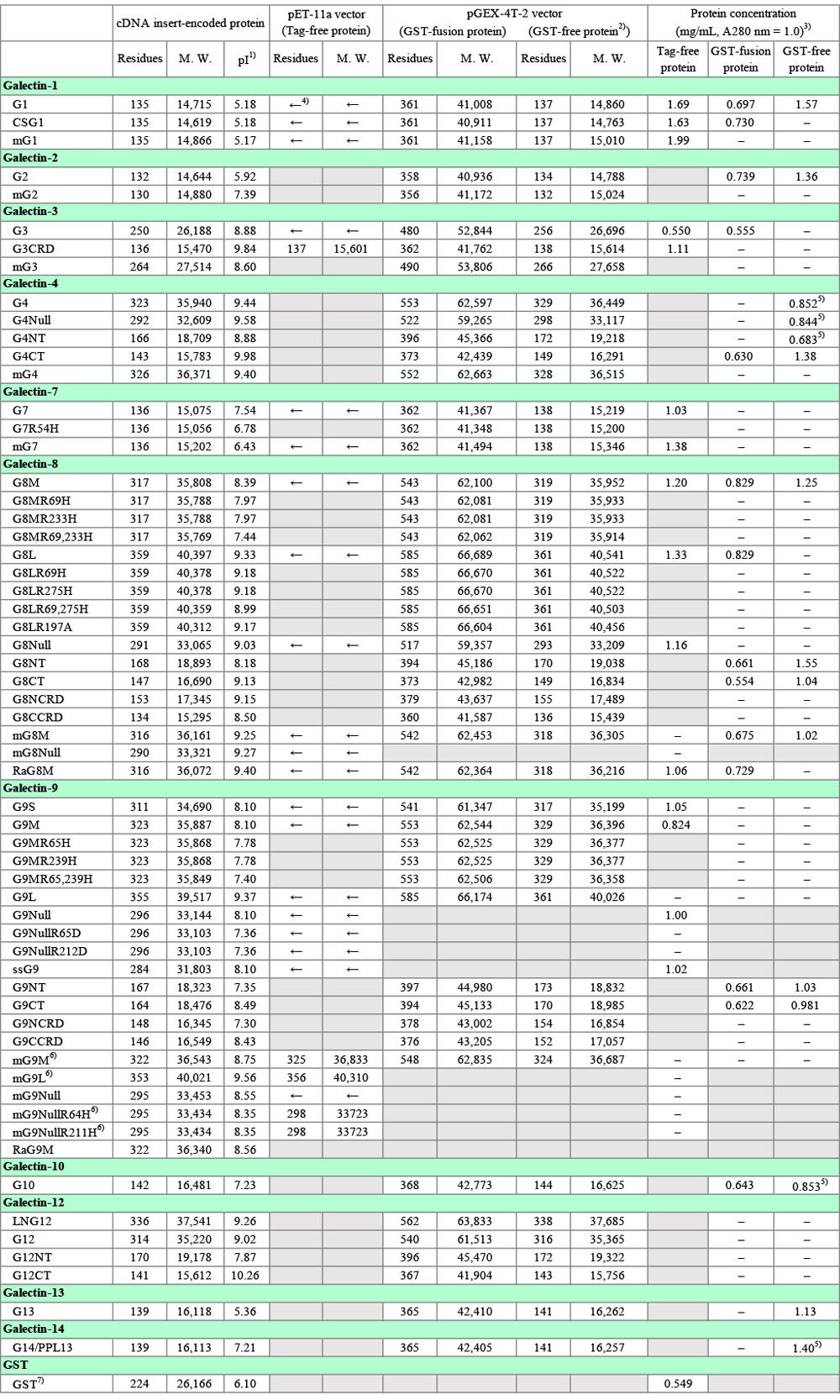
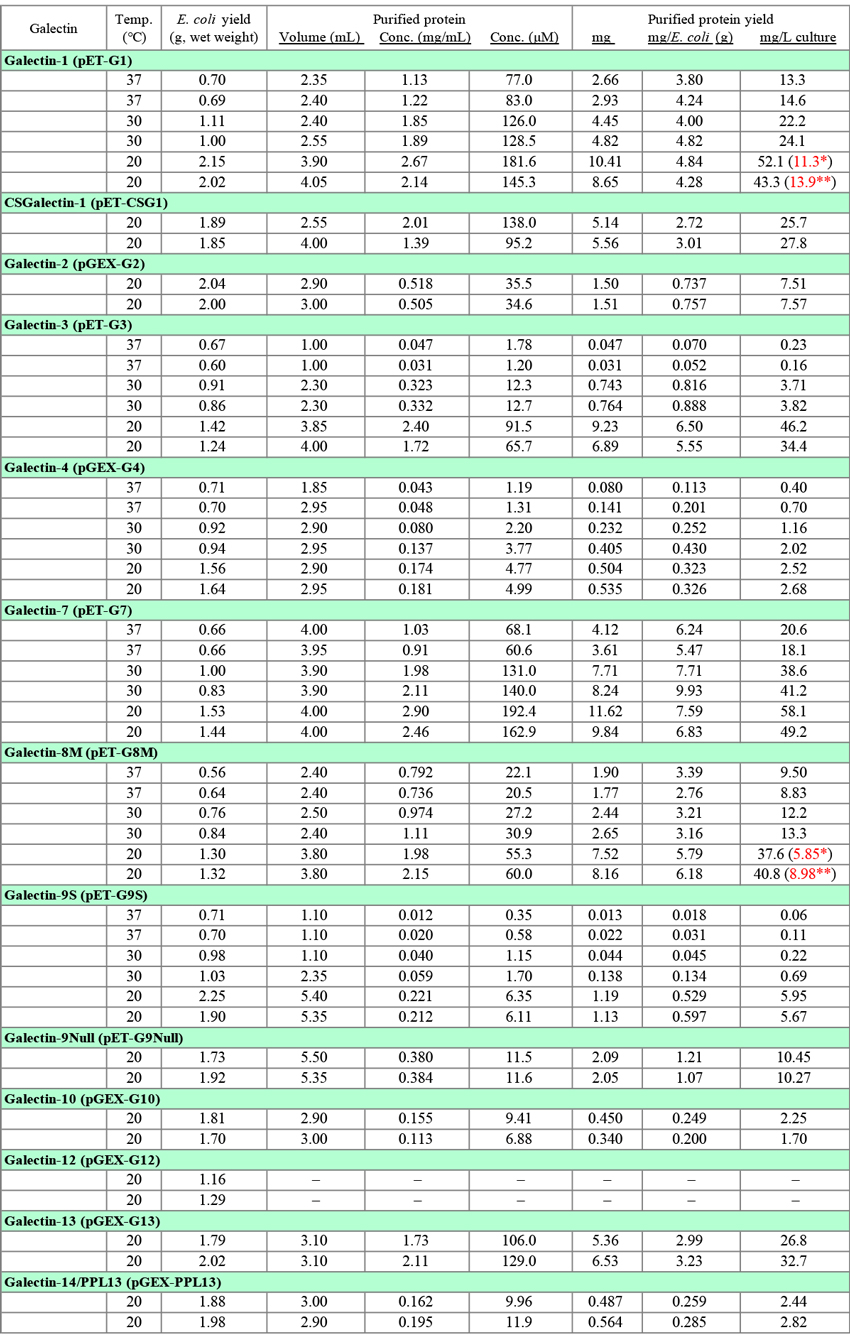
Table 3は、大腸菌を宿主とした組換えタンパク質の生産(収量)に関するデータです。ガレクチンの発現実験はこれまでに数多く行ってきましたが、実験条件が同一ではないため、このノートを作成するにあたってデータを取り直しました。G1, G3, G4, G7, G8M, G9Sについては、3種類の発現条件(37°C, 2 h; 30°C, 3 h; 20°C, 16 h)、それ以外は単一条件(20°C, 16 h)で各2回行った実験データをそのまま記載しました。GST融合タンパク質(G2, G4, G10, G13, G14/PPL13)の場合、GSTタグを除去したものを精製タンパク質としました(Fig. 2参照)。発現・精製条件をFig. 1(pET vector)とFig. 2(GST vector)に示します。Fig. 1の内容はGlycoPOD(GlycoScience Protocol Online Database; Sugar binding proteins: Expression and purification of recombinant human galectin-9)に記載したものと基本的に同じですが、一部変更があります。なお、このノートでは大腸菌以外の宿主については触れませんが、ガレクチンファミリーの中で最も発現が困難なメンバーの1つであるG9の場合、これまでに試みた宿主(大腸菌、酵母 [S. cerevisiae, P. pastoris]、バキュロウイルス感染昆虫細胞 [expres SF+]、哺乳動物細胞 [COS-1/7, HEK293, CHO]、コケ [Physcomitrella patens])の中では、大腸菌が最も優れていることが分かっています。また、codon optimizationもG9の発現には効果がありませんでした。大腸菌以外の宿主に関するデータの大部分は、(株)ガルファーマ(外注を含む)において得られたものです。
現在、ガレクチンの発現はすべて20°C, overnight(14 - 18 h)で行っています。Table 3の精製タンパク質の収量データ(mg, mg/L culture)から分かるように、発現温度と時間を変えて収量を比較したすべてのガレクチンで、20°C, 16 hの条件が最も高収量となっています。しかし、収量を菌体湿重量あたり(mg/E. coli [g])で比較すると、G1やG7では発現温度による差はほとんど認められず、精製タンパク質の収量は菌体収量の違いを反映していることを示しています。一方、G3とG9Sの場合、20°C以外では収量が極端に低く、十分な収量を得るためには低温での発現が必須と考えられます。Table 3のデータは小スケールの発現実験で得られたもので、一般的には、スケールアップにより収量(mg/L culture)は増加します。
組換えタンパク質の各精製段階における SDS-PAGEのパターン(後述)を調べると、菌体抽出液をlactose-agaroseでバッチ処理した後の遠心上清(非吸着画分)や、カラムの洗浄画分に相当量の組換えタンパク質が含まれている場合があります。G1とG8Mについて、非吸着画分と洗浄画分から組換えタンパク質を回収したデータをTable 3に赤字で示してあります*。かなりの量を回収することができますが、操作には手間がかかりますので、十分な収量が得られている場合、回収操作を行うメリットは少ないと思います。洗浄画分へのロスに関しては、洗浄用緩衝液の量を減らすことで、最終標品の純度を落とすことなく、ある程度防ぐことができると思われます。glutathione-Sepharoseの場合、少なくともカラムの洗浄画分への組換えタンパク質の漏出はほとんどありません。
* 非吸着画分は菌体抽出液と同様にバッチ法で、洗浄画分は1 mLのゲルをパックしたカラムを用いて精製した。ゲルの洗浄、溶出、透析などの条件はFig. 1(Step 18/19 - 24)と同じ。
Procedure
1) Inoculate 10 mL of LB-broth containing 100 µg/mL ampicillin with a loop full of frozen glycerol stock BL21(DE3)* cells carrying the expression plasmid.
* In the case of pET vectors, an expression host containing a chromosomal copy of the T7 RNA polymerase gene under lacUV5 control, e.g., E. coli BL21(DE3), must be used.
↓
2) Incubate the inoculum at 37°C overnight with shaking (200 rpm) in a shaking incubator.
↓
3) Dilute 4.4 mL of the overnight culture with 220 mL of 2xYT medium containing 100 µg/mL
ampicillin in a 1,000-mL flask.
↓
4) Incubate the inoculum at 37°C for ~ 2 h with shaking until the absorbance at 600 nm (A600 nm)
reaches 0.6 - 0.7*.
* Measurements of absorbance were done on samples collected at intervals by withdrawing
culture liquid from the flask.
↓
5) Add 0.22 mL of 0.1 M IPTG (isopropyl-β-thiogalactopyranoside) solution to the culture
(final concentration, 0.1 mM).
↓
6) Incubate the culture at 37°C for 2 h/at 30°C for 3 h/at 20°C for 16 h (overnight) with shaking
(200 rpm).
↓
7) Transfer 200 mL of the overnight culture to centrifuge tubes and then spin down the cells at
6,000 xg for 10 min.
↓
8) Resuspend the pellet in 36 mL of 10 mM Tris-HCl (pH 7.5), 0.5 M NaCl, 1 mM PMSF
(phenylmethylsulfonyl fluoride)*
* 10 mM Tris-HCl (pH 7.5), 0.5 M NaCl, 1 mM PMSF, 1 mM DTT (dithiothreitol) was used
for G1 and mG1.
↓
9) Transfer the cell suspension to a cooling cell and then lyse the E. coli cells using a sonicator*.
* When using a rosette cooling cell and a Sonifier 250 (Branson) equipped with a 1/2" diameter horn, sonicate the cell suspension for 2min x 2 and an additional 1 min (with 1-min intervals between the bursts) with the following settings: % duty cycle, 100; output control, 5.0. Place the cooling cell in an ice-water bath during the sonication.
↓
10) Add 4 mL of 10% (w/v) Triton X-100 to the cell lysate (final concentration, 1%).
↓
11) Mix the lysate for 30 min at 4°C with a magnetic stirrer.
↓
12) Transfer the lysate to a centrifuge tube and then spin down the cell debris at 18,000 xg for
30 min at 4°C.
↓
13) Transfer the supernatant to a 100-mL bottle.
↓
14) Add 2 mL of the lactose-agarose slurry (50%[v/v] in TBS [Tris-buffered saline: 20 mM Tris-HCl, pH 7.5, 0.15 M NaCl]) to the supernatant.
↓
15) Mix the lactose-agarose gel suspension by rotating the bottle for 1 h on a tube rotator*.
* Do not use a magnetic stirrer to avoid destruction of the gel beads.
↓
16) Transfer the gel suspension to a 50-mL conical tube and then spin down the gel at 1,500 xg for 5 min.
↓
17) Discard the supernatant and then suspend the gel pellet in an appropriate volume of TBS, 0.03% CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate).
↓
18) Pack the gel into a column.
↓
19) Wash the gel with 20 gel-bed volumes of TBS, 0.03% CHAPS.
↓
20) Elute the recombinant protein from the gel with 4 mL (1 mL x 4) of TBS, 0.2 M lactose.
↓
21) Dialyze the eluate against PBS*.
* 1st, 4 - 5 h (500 mL); 2nd, overnight (1,000 mL); 3rd, 4 - 5 h (500 mL)
↓
22) Transfer the dialyzed solution to a centrifuge tube, and then spin down the insoluble material at
25,000 xg for 20 min.
↓
23) Sterilize the supernatant with a sterile filter (0.2 µm).
↓
24) Store the sterilized solution at 4 °C.
Procedure
1) Inoculate 10 mL of LB-broth containing 100 µg/mL ampicillin with a loop full of frozen
glycerol stock BL21 cells carrying the expression plasmid.
↓
2) Incubate the inoculum at 37°C overnight with shaking (200 rpm) in a shaking incubator.
↓
3) Dilute 4.4 mL of the overnight culture with 220 mL of 2xYT medium containing 100 µg/mL ampicillin in a 1,000-mL flask.
↓
4) Incubate the inoculum at 37°C for ~ 2 h with shaking until the absorbance at 600 nm (A600 nm) reaches 0.6 - 0.7*.
* Measurements of absorbance were done on samples collected at intervals by withdrawing culture liquid from the flask.
↓
5) Add 0.22 mL of 0.1 M IPTG solution to the culture (final concentration, 0.1 mM).
↓
6) Incubate the culture at 37°C for 2 h/at 30°C for 3 h/at 20°C for 16 h (overnight) with shaking (200 rpm).
↓
7) Transfer 200 mL of the overnight culture to centrifuge tubes and then spin down the cells at 6,000 xg for 10 min.
↓
8) Resuspend the pellet in 36 mL of 10 mM Tris-HCl (pH 7.5), 0.5 M NaCl, 1 mM PMSF*
* 10 mM Tris-HCl (pH 7.5), 0.5 M NaCl, 1 mM PMSF, 1 mM DTT was used for G1 (GST-G1) and mG1 (GST-mG1).
↓
9) Transfer the cell suspension to a cooling cell and then lyse the E. coli cells using a sonicator*.
* When using a rosette cooling cell and a Sonifier 250 (Branson) equipped with a 1/2" diameter
horn, sonicate the cell suspension for 2 min x 2 and an additional 1 min (with 1-min intervals between the bursts) with the following settings: % duty cycle, 100; output control, 5.0. Place the cooling cell in an ice-water bath during the sonication.
↓
10) Add 4 mL of 10% (w/v) Triton X-100 to the cell lysate (final concentration, 1%).
↓
11) Mix the lysate for 30 min at 4°C with a magnetic stirrer.
↓
12) Transfer the lysate to a centrifuge tube and then spin down the cell debris at 18,000 xg for
30 min at 4°C.
↓
13) Transfer the supernatant to a 100-mL bottle.
↓
14) Add 2 mL of the glutathione-Sepharose slurry (50%[v/v] in TBS) to the supernatant.
↓
15) Mix the glutathione-Sepharose gel suspension by rotating the bottle for 1 h on a tube rotator*.
* Do not use a magnetic stirrer to avoid destruction of the gel beads.
↓
16) Transfer the gel suspension to a 50-mL conical tube and then spin down the gel at 1,500 xg for
5 min.
↓
17) Discard the supernatant and then suspend the gel pellet in an appropriate volume of TBS,
0.03% CHAPS.
↓
18) Pack the gel into a column.
↓
19) Wash the gel with 20 gel-bed volumes of TBS, 0.03% CHAPS.
↓
20) Wash the gel with 2 gel-bed volumes of PBS.
↓
21) Add 1 mL of thrombin solution (50 u/mL in PBS) to the gel in the column, and then mix.
↓
22) Incubate the gel suspension (in the column) at 25°C for 16 h (overnight) with shaking (120 rpm).
↓
23) Drain the GST-free recombinant protein solution from the column (eluate-1).
↓
24) Elute the GST-free recombinant protein from the gel with 2 mL (1 mL x 2) of PBS (eluate-2).
↓
25) Dialyze the combined eluates (eluate-1 and eluate-2) against PBS*.
* 1st, 4 - 5 h (500 mL); 2nd, overnight (1,000 mL); 3rd, 4 - 5 h (500 mL)
↓
26) Transfer the dialyzed solution to a centrifuge tube, and then spin down the insoluble material at
25,000 xg for 20 min.
↓
27) Sterilize the supernatant with a sterile filter (0.2 µm).
↓
28) Store the sterilized solution at 4 °C.
・Step 1
GST Gene Fusion Systemのマニュアル(GST Gene Fusion System Handbook 18-1157-58 AD 11/2014, GE Healthcare Life Sciences;以下、GSTマニュアル)では、この段階から培地として2xYT(+ ampicillin)を使用しています。
・Step 3
今回の発現実験では、1,000 mLのフラスコに220 mLの培地(+ overnight culture)を入れて培養を行っています。同じフラスコで400 mL程度の培地を用いても、収量(mg/L culture)に大きな違いはありませんでした。但し、この発現実験ではフラスコの栓(カバー)として2枚重ねのアルミ箔を使っていますので、通気性の低い培養栓を用いた場合、収量に差が出る可能性があります。
GSTマニュアル:overnight cultureを発現用培地で1:100に希釈。
・Step 4
吸光度*をチェックするための培地のサンプリングは、フラスコをインキュベーターシェーカーにセットしたまま行っていますので、完全に無菌的な条件ではありません。IPTG(isopropyl β-D-1-thiogalactopyranoside)を添加後、30°Cあるいは20°Cで培養を行う場合、吸光度が0.5程度になった時点で、装置の設定温度を下げています。ほとんどの実験で600 nmにおける吸光度が0.60 - 0.65になったタイミングでIPTGを加えていますが、一般的な分光光度計で大腸菌培養液の吸光度を測定した場合、測定値が安定しないため、厳密な値とはいえません。
* 実際には吸収ではなく、大部分は測定光の散乱によるものです。
GSTマニュアル:A600 nm = 0.5 ~ 2でIPTG添加(最終濃度0.1 ~ 1 mM)。
・Step 8
菌体懸濁用緩衝液として10 mM Tris-HCl (pH 7.5), 0.5 M NaCl, 1 mM PMSF(phenylmethanesulfonyl fluoride, synonym: benzylsulfonyl fluoride)*を使用していますが、ガレクチン用に最適化したものではありません。スケールアップする場合は、同じ割合(培養液の18%、Triton X-100を加えて20%)の緩衝液を使用しています。PMSFは水中では不安定です。DMSO(dimethylsulfoxide)に溶解した場合でも、DMSOに含まれる水分によって分解が進みますので、用時調製が原則です。
* G1/mG1の場合は、さらに1 mM DTT(dithiothreitol)を添加(G1の項参照)。DTTは、G1/mG1のレクチン活性以外に、組換えタンパク質の純度や収量にも影響を与える可能性がありますが、この点についての検討は行っていません。
GSTマニュアル:菌体懸濁用緩衝液として、培養液100 mLに対して5 mLのPBSを使用。
・Step 9
音波処理の際、冷却効率を上げるためにロゼットクーリングセル(Rosette Cooling Cell; Fig. 3A)を氷水に浸けた状態で使用しています。音波処理に伴う温度上昇が最終標品の収量や活性にどの程度影響するのかは不明ですが、冷却効率の悪い容器を使用する場合、1回の処理時間を短くするなどの変更が必要かもしれません。
・Step 10
組換えタンパク質の可溶化を促進する目的でTriton X-100(最終濃度1%)を加えています(GSTマニュアルと同じ条件)。菌体懸濁用緩衝液と同様、習慣的に使用しているもので、ガレクチンの精製に関して十分な検討は行っていません。
・Step 14
通常、400 - 500 mLの培養液に対して1.5 mLのゲルを使用しています。バッチ法で吸着を行う場合、その後のステップでのロスを考えるとゲルの量が少なすぎるのは問題なので、この実験では200 mLの培養液に対して1 mLのゲルを使用しました。手元にあるlactose-agarose(ホーネンコーポレーション)の製品添付文書には、結合容量としPeanut agglutinin (molecular weight = 98,000), 5.0 mg/mL gel(= 51 nmol/mL gel)以上、と記載されています。Table 3の精製タンパク質収量(mg)から計算すると、G1の場合(20°C, 16 h)約600 nmol(ダイマーを1分子とすると約300 nmol)、G8Mの場合は約200 nmolが1 mLのゲルを使って精製されたことになります。前述のように、非吸着画分と洗浄画分に相当量の組換えタンパク質が含まれる場合があることを考慮すると、発現量の多いガレクチンの場合、ゲルの量が不十分である可能性は否定できません。GSTマニュアルでは、400 mLの培養液あたり0.2 mLのglutathione-Sepharoseゲルを使用(但し、バッチ法ではなくカラム法を推奨;GST-tagged proteinの収量を1 mgと想定)していますが、やや少なすぎるように感じます。glutathione-Sepharose 4Bの結合容量は、製品添付文書ではGST, > 5 mg/mL gel、GSTマニュアルでは、horse liver GST, 25 mg/mL gelと記載されています。
吸着法としてバッチ法を用いているのは、他の組換えタンパク質を精製する際に、菌体抽出液の粘度が高く、実質的にカラム法が使えない場合があったためです。但し、ガレクチンの発現実験で抽出液の粘度が高くなることは経験していません。
・Step 15
Fig. 3B参照。
・Step 18
Econo-Column Chromatography Column(1.0 x 10 cm, Bio-Rad)を使用しています。ゲルのパッキングと洗浄時は、流速を上げるためにカラムの出口に長さ10 cm程度のチューブを着けています(Fig. 3C;溶出時にはチューブを外す)。
・Step 19
洗浄用緩衝液は0.03% CHAPS(3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate)を含んでいます。両性(両イオン性)界面活性剤であるCHAPSは、タンパク質を扱う上で様々な利点を持っているため好んで使用していますが 1、比較的高価なことが欠点です。界面活性剤を含まない、あるいはTween 20などの非イオン性界面活性剤*を含む洗浄液を使用しても、精製標品の純度への影響は少ないと思います。
* 一般的に、非イオン性界面活性剤には、1) 透析で除きにくい(臨界ミセル濃度が低く、ミセル量が大きいため)、2) 細胞毒性が高い、3) 280 nmに強い吸収を持つことが多い、などの問題があります。非イオン性界面活性剤を使用した場合、溶出前に界面活性剤を含まない緩衝液でゲルを洗浄する必要があります。
・Step 20(Fig. 1)
lactose-agaroseゲルの場合、溶出液として習慣的にTBS, 0.2 M lactoseを使用しています。ガレクチンファミリーの中でlactoseに対して比較的高い親和性を示すG9は、10 - 15 mM lactoseでlactose-agaroseから溶出されますので(精製したG9をlactose-agaroseに結合させた後、グラジエント溶出を行った場合の溶出ピークのlactose濃度)、溶出液のlactose濃度を50 - 100 mM程度まで下げても問題ないと思います。galactoseはlactoseと較べて高価なのでメリットは少ないですが、溶出剤として500 mM galactoseを使うことも可能です。G9は200 - 250 mM galactoseで溶出されます(上述の条件)。ガレクチンのX線結晶解析に際して、精製過程でlactoseを使用した場合、十分に透析を行っても一部のガレクチン分子に結合したlactoseが結晶中に検出されることがあります 2。確認は行っていませんが、lactoseの代わりにgalactoseを使うことで、糖をほとんど含まない結晶が得られる可能性があります。
精製に使用したゲル(lactose-agarose, glutathione-Sepharose)は、再生して繰り返し使用しています。使用後のゲルに防腐剤(0.05% NaN3など)を加えて4°Cで保存し、ある程度溜まった(~ 20 mL)ところでまとめて再生します。ゲルをカラム(Econo-Column Chromatography Column, 2.5 x 10 cm)に充填し、ゲルベッドの2倍量のPBS、2倍量の6 M guanidine hydrochloride水溶液、5倍量のPBS、の順番で洗浄し、最終的に50% (v/v) slurryとなるようにPBS, 0.05% NaN3に懸濁して4°Cで保存しています。再生回数は特に限定していません。Fig. 2(Step 21以降)に示したように、GST融合タンパク質がゲルに結合した状態でthrombin処理を行った場合は、GST-freeの組換えタンパク質を回収した後、ゲルを保存する前に50 mM Tris-HCl (pH 8.0), 10 mM glutathioneを使ってglutathione-Sepharoseに結合しているGSTを溶出(1 mL x 4、各溶出の間に5分間のインターバル)しています。
・Step 21(Fig. 1)
透析の条件(透析外液の量、時間、交換回数)も習慣的に用いているものです。おそらく、外液の量を減らし、短時間で外液を交換する(交換回数は増やす)ことで、より効率良くlactoseなどを除くことができると思われますが、裏付けとなるデータは持っていません。
・Step 24(Fig. 1)
精製標品の安定性については各ガレクチンの項で触れますが、一般的にタンデムリピート型ガレクチン(Nullタイプの改変体を除く)は、比較的短期間で分解されます(混入しているプロテアーゼによるリンカーペプチドの切断)。また、不溶化する可能性があるため、凍結保存する際は注意が必要です。
・Step 20(Fig. 2)
Fig. 2ではglutathione-Sepharoseゲルに結合した状態でthrombin処理を行っています(Step 21以降)。GST融合タンパク質としてそのまま使用する、あるいは融合タンパク質を溶出した後にthrombin処理を行う場合は、このステップで50 mM Tris-HCl (pH 8.0), 10 mM glutathioneを使って溶出します。GST融合タンパク質を溶出する際の注意点については、GSTマニュアルを参照して下さい。
・Step 21, 22(Fig. 2)
ボルテックスミキサーを使って、カラム内でゲルとthrombin溶液を混合しています。overnight処理の間も、十分な混合効果が得られるわけではありませんが、出口のコックを閉じ、上部をパラフィルムで覆ったカラムをインキュベーターシェイカーで撹拌しています。今回、一律にovernight処理を行いましたが、数時間で反応がほぼ完了する場合もあります。大腸菌由来のプロテアーゼによる分解が問題となる場合は、時間を短縮するなど(必要に応じてthrombinを増量する)、処理条件の検討が必要です。
・Step 23 ~ 27(Fig. 2)
ステップ23, 24で得られるGST-free組換えタンパク質を含む溶液の吸光度(A280 nm)はかなり高い値を示しますが、透析・遠心後の上清の吸光度は大幅に低下します。以下はG4の例(20°C, 16 h)です。supernatantはeluate-1とeluate-2 (1st & 2nd) を混合して透析した後、遠心、沪過滅菌したサンプルです。
| Sample | Volume (mL) | A280 nm |
| eluate-1 | 1.0 | 1.346 |
| eluate-2 (1st) | 1.0 | 1.241 |
| eluate-2 (2nd) | 1.0 | 0.348 |
| supernatant | 2.9 | 0.204 |
各サンプルのSDS-PAGEデータは、透析・遠心や沪過滅菌によるロスではこの差を説明できないことを示しています(data not shown)。eluate-1, -2の紫外部吸収スペクトルのピークがおよそ260 nm、supernatantは280 nmであることを考え合わせると、eluate-1, -2は核酸(透析で除去可能な核酸)を含んでいる可能性が高いと考えられます。他のGST融合タンパク質の場合も同様です。
最終標品はthrombinを含んでいます。必要に応じてbenzamidine-SepharoseやHiTrap benzamidine FF (HS)カラム(GE Healthcare Life Sciences)を用いてthrombinを除くことができますが(GSTマニュアル参照)、benzamidineは中性付近のpHでは正に荷電(酸解離定数:11.90 ± 0.40)しているため、注意が必要です。等電点が低いG2(pI = 5.92)の場合、Fig. 2の方法で得られたeluate-1, -2(PBS溶液)をそのままHiTrap benzamidineカラムにアプライすると、thrombinだけでなくG2(GST-free G2)も担体に結合します(Fig. 4D AFT;図の説明は下記参照)。予めNaCl濃度を0.5 Mに上げておくことで(例えば、1 mLのPBS溶液あたり約0.08 mLの5 M NaClを加える)、この問題を回避できます(Fig. 4D BFT)。
eluate-1 + eluate-2(この実験はFig. 2の2倍のスケールで行ったため、約6 mL)を2等分し、一方(A)はそのまま、他方(B)には5 M NaClを加えてNaCl濃度を0.5 Mに調整し、benzamidine固定化カラム(HiTrap benzamidine FF(HS), 1 mL)に流した(流速、0.5 mL/min)。カラムの平衡化緩衝液として、Aサンプルの場合はPBS、Bサンプルの場合は20 mM Na-Pi (sodium phosphate) (pH 7.5), 0.5 M NaClを使用した。非吸着画分(素通り画分)を保存し、カラムに結合した成分を、Aサンプルの場合は20 mM Na-Pi (pH 7.5), 0.5 M NaClと0.1 M Glycine-HCl (pH 2.7)(各2 mL)、Bサンプルの場合は0.1 M Glycine-HCl (pH 2.7)(2 mL)で溶出した。染色バンドを直接比較できるように、各レーンにアプライするサンプル量を調整してある。なお、Fig. 4 - 7 & 9に示すSDS-PAGEでは2種類の分子量マーカー(Unstained Protein Standard, Broad Range [10-200 kDa], P7704 & P7717, New England BioLabs)を使用しています(Fig. 4Dの場合はP7717)。
| G2: | eluate-1 + eluate-2 |
| AFT, BFT: | 非吸着画分 |
| AE1: | 20 mM Na-Pi (pH 7.5), 0.5 M NaCl 溶出画分 |
| AE2, BE2: | 0.1 M Glycine-HCl (pH 2.7) 溶出画分 |
GSTマニュアル:

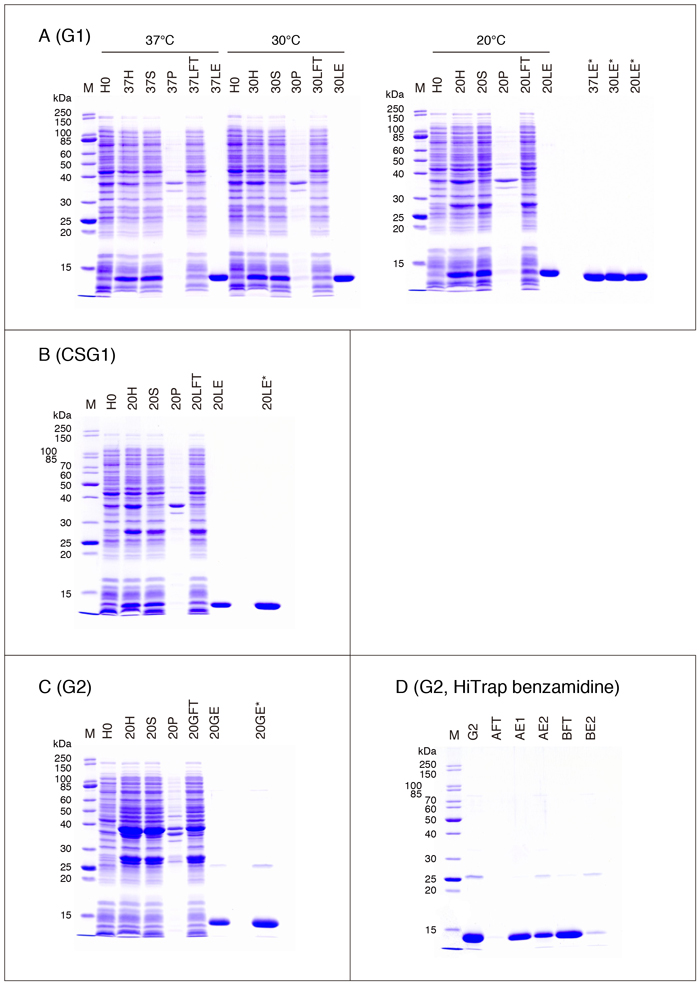
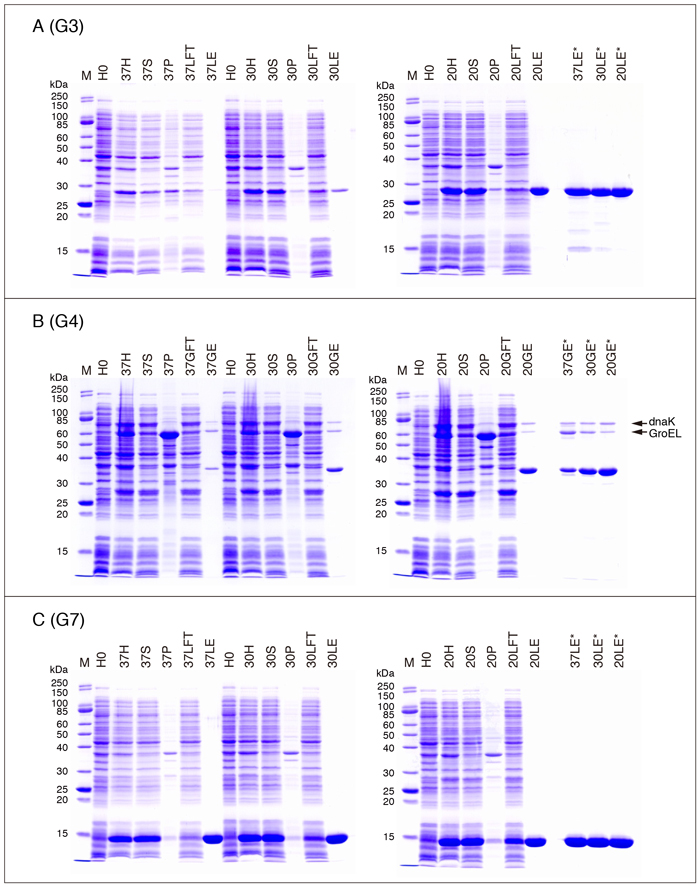
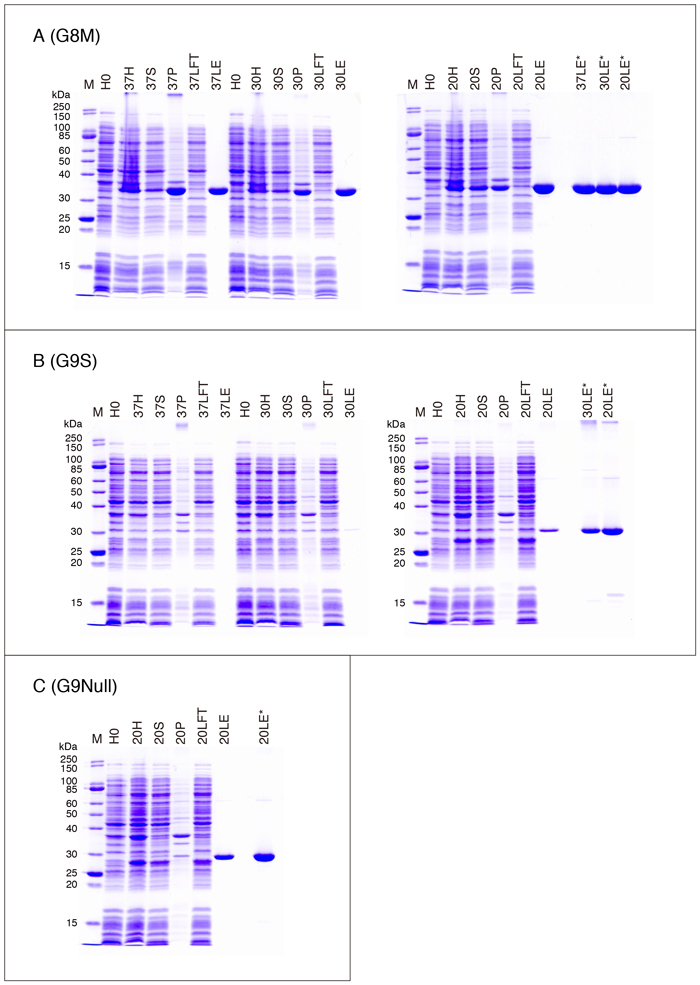
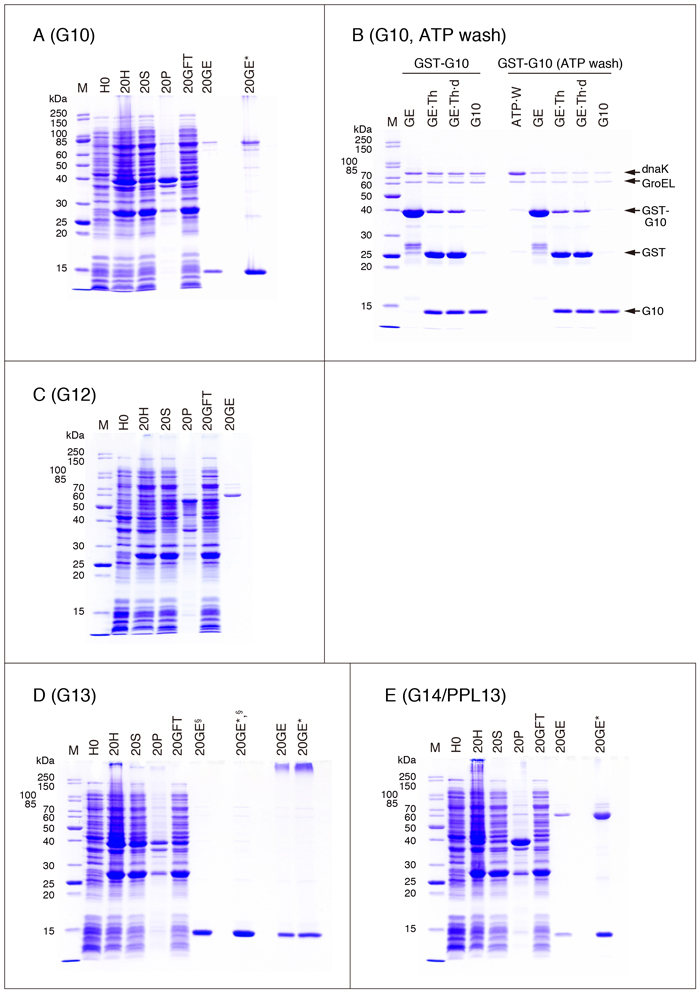
Fig. 4A - C, Fig. 5A - C, Fig. 6A - C, Fig. 7A, C, D, Eに、各ガレクチンの発現・精製過程におけるSDS-PAGEパターンを示します。Table 3のデータを得た際のサンプル(2回行った実験の何れか一方)を使用しています。以下は泳動サンプルについての説明です。
| H0: | IPTGを加える直前のサンプル(Step 4) |
| H(37H/30H/20H;以下同様): | 菌体懸濁液を音波処理・Triton X-100処理した後のサンプル*(Step 11) |
| S: | Triton X-100処理後の遠心上清(Step 12) |
| P: | Triton X-100処理後の遠心沈渣(Step 12) |
| LFT: | lactose-agaroseバッチ処理後の遠心上清(Step 16) |
| GFT: | glutathione-Sepharoseバッチ処理後の遠心上清(Step 16) |
| LE: | 最終精製標品(purified G1 etc.)(Step 23, Fig. 1) |
| GE: | 最終精製標品(purified G2 etc.)(Step 27, Fig. 2) |
| 泳動サンプル量: | H0 - LFT/GFT, 0.1 mg wet pellet equivalent**/10 µL (10 µL/lane) LE/GE (= purified protein), ガレクチンにより異なる(図の説明に記載) |
* 理由は不明ですが、Triton X-100処理を行う前のサンプル(Step 8, 9)を用いた場合、ガレクチンの種類によっては、SDS-PAGEで発現タンパク質のバンドが確認できないことがあります。
** H - LFT/GFTの場合、1レーンあたりの泳動量は、Step 7で得られたE. coliペレットの湿重量(Table 3, E. coli yield)0.1 mgに相当/由来する(0.1 mg wet pellet equivalent)サンプルです。例えばG1の場合(37°Cの1回目)、菌体湿重量は0.7 gなのでStep 10/11では0.7 g E. coli/40 mL(36 mL [菌体懸濁用緩衝液] + 4 mL [10% Triton X-100];菌体沈渣による体積の変化は無視する)→ 17.5 µg wet E. coli pellet/µL (17.5 µg wet pellet equivalent/µL)となります。下記は0.1 mg wet pellet equivalent/10 µLの泳動サンプルを100 µL作製する場合の条件です。
Step 11のサンプル, 57.1 µL + SDS sample buffer (4X)*, 25 µL + H2O, 17.9 µL
* 0.25 M Tris-HCl (pH 6.8), 8%(w/v) SDS, 20%(v/v) mercaptoethanol, 25%(w/v) glycerol
H0の場合は、Step 6で得られる培養液(overnight culture)のA600 nmの値を測定し、菌体湿重量と培養液のA600 nmの関係を計算します。上述のG1の例では、A600 nm = 1.0の培養液1 mLから得られる菌体湿重量は2.38 mgとなります。この値はクローンによってかなりのばらつきがあります(50個のデータの平均と標準偏差:1.93 ± 0.55 mg)。IPTGを加える直前にサンプリングした培養液(H0)はA600 nm = 0.63であったため、前述の関係を当てはめると、この培養液1 mLを遠心して得られる菌体ペレットの湿重量は、1.50 mgとなります*。このペレットに150 µLのSDS sample buffer (1X)を加えて処理すれば、0.1 mg wet pellet equivalent/10 µLの泳動サンプルが得られることになります(菌体沈渣による体積の変化は無視する)。Step 4に関するコメントの中で触れたように、一般的な分光光度計で大腸菌培養液の吸光度を厳密に測定することは困難です。また菌体密度とA600 nmの値は、菌体密度が高くなると比例しなくなりますので、Step 6の培養液の場合、測定値が概ね0.7以下になるように希釈して測定しています。
* 菌体ペレット(H0サンプル)は、菌体湿重量と培養液(overnight culture)のA600 nmの関係が計算できるまで凍結保存。
・G1
ガレクチンは当初、レクチン活性の発現に還元型のシステイン残基(-SH基)が必要なレクチンファミリーと考えられ、C型レクチンとの対比でS型レクチンと呼ばれていました(この呼称は現在でも使われています)。よく知られているように、これはG1がシステイン残基の酸化(分子内/分子間ジスルフィド結合の形成)に伴う構造変化によってレクチン活性を失うためであって、-SH基が糖鎖との結合に必須なためではありません 3。G1/mG1の場合、Step 8(Fig. 1, 2)では1 mM DTTを添加した菌体懸濁用緩衝液を使用していますが、最後に使用する透析外液には還元剤を加えていません。このままの状態で保存すると、G1の場合、10日後には活性(赤血球凝集活性)は約1/4に低下します 4。最終標品に1 mM DTTを加えた場合でも、20日後には同程度まで低下します。赤血球凝集活性は、レクチン活性の指標として広く利用されている鋭敏な測定方法です。一般的に、培養細胞に対するガレクチンの作用発現には、赤血球凝集活性と較べてより高い濃度を必要としますので、赤血球凝集活性を検出可能な標品でも、培養細胞などに対する作用が実質的に失われている場合があります(G7, G8の項参照)。
CSG1はG1の全てのCys残基をSer残基に置換した変異体で、還元剤が存在しない条件下で長期間保存しても活性は低下しません 4。この変異は立体構造や糖鎖結合特異性に影響を与えませんので、CSG1をG1の代替物として利用することができます。G1はレクチンとして以外に、神経栄養因子としての機能を持つことが知られています。分子内で3本のジスルフィド結合を形成したoxidized G1は、レクチン活性を失う一方で、マクロファージなどを介した末梢神経の再生促進活性を獲得することが報告されています 5。従って、酸化によって神経栄養因子としての機能を獲得する可能性がないという点で、CSG1はG1の完全な代替物ではありません。
・G2
G2はlactoseに対して親和性を示さないわけではありませんが 6,7、lactose-agaroseを用いたアフィニティー精製は実質的に不可能です。なお、文献6, 7ではPA-化(pyridylamination)あるいはpNP-化(p-nitrophenylation)された糖鎖を用いたFAC(frontal affinity chromatography)によって親和性を測定していますので、アガロースゲルに固定化されたlactoseの場合は多少異なる可能性があります。精製G2標品(Fig. 4C)に含まれている分子量約26,000のバンドは、切断されたGST-tagの混入と考えられます(Fig. 7B参照)。
・G3
Fig. 5Aの右側に各発現温度での精製G3標品(3 µg/10 µL [10 µL/lane])の泳動パターンを並べて示してあります。この実験で用いた精製標品の濃度は47 µg/mL (37°C), 323 µg/mL (30°C), 2.40 mg/mL (20°C)なので、SDS sample buffer (4X)を使用した場合、発現温度が20°Cの標品以外は3 µg/10 µLの泳動サンプルを作製することはできません。そのため、37°C/30°Cの標品は、SDS処理する前にSCR(StrataClean Resin)を使って濃縮しています。SCRに関する説明は「補足」の項を参照して下さい。
G3はキメラ型サブファミリーに属する唯一のガレクチンで、N-末端側の非レクチンドメイン(コラーゲン様ドメイン)とC-末端側CRDから構成されています。G3CRD(Ile115 - Ile250)はこのC-末端側CRDに相当します*。Table 3とは条件が異なりますが、以前に行った実験(pET-G3CRD; 20°C overnight)における精製標品の収量は約3 mg/L cultureで、G3の1/10以下でした。
* pET-G3CRDの産物はN-末端側にベクター配列由来のMetが付加される。
・G4
G4をlactose-agaroseを用いてアフィニティー精製することはG2と同様、実質的に不可能です。
G3の場合と同じように、G4の精製標品(G4の場合は全ての温度)もタンパク質濃度が低いため、SDS処理する前にSCRを使って濃縮しています(Fig. 5B, 37GE*/30GE*/20GE*)。濃縮した3つの精製標品でバンドの濃さは同じになるはずですが、明らかに異なっています。これは、サンプル(特に発現温度が37°Cと30°Cの精製標品)がタンパク質以外の不純物(おそらくは核酸)を含んでいるため、A280 nmから計算したタンパク質濃度が実際よりも高くなっているためと思われます。精製標品に含まれている分子量約76,000と63,000のバンドは、各々大腸菌シャペロンのDnaKとGroELです(各バンドのN-末端アミノ酸配列を確認*)。G4が結合した状態のglutathione-SepharoseをATP溶液で洗浄することによって、DnaKをある程度除くことができます(G10の項参照)。
* 分子量約76,000のバンド:GKIIGIDLGT
分子量約63,000のバンド:AAKDVKFGNDARVKM
G4Null, G4NT, G4CTの構造は以下のとおりです。
| G4Null: | N-末端側CRD(Met1 - Gln153)+ リンカーペプチドの一部 (Pro154-Leu155 + Thr187 - Pro191)+ C-末端側CRD(Val192 - Ile323) |
| G4NT: | N-末端側CRD(Met1 - Gln153)+ リンカーペプチドの一部(Pro154 - Tyr166) |
| G4CT: | リンカーペプチドの一部(Thr181 - Pro191)+ C-末端側CRD(Val192 - Ile323) |
1度だけの実験データですが(Table 3とほぼ同じ条件)、G4Null, G4NT, G4CT(GST-tagを除いた精製標品)の収量は、各々、約1.6 mg/L culture, 10 mg/L culture, 20 mg/L cultureでした。
・G7
G7R54Hは、糖鎖結合サイトを形成する重要なアミノ酸残基の1つであるArg54をHisに置換した変異体です。ほぼ糖鎖結合活性を失っていますが、赤血球凝集活性は検出できる可能があります。他のガレクチンの場合も同様です(G8の項参照)。
・G8
ヒトG8には、リンカーペプチドの構造のみが異なる少なくとも2種類のアイソフォーム(スプライシングアイソフォーム:G8M, G8L)が存在します。G8MとG8Lのリンカーペプチドは、28残基と70残基です。G8L(pET-G8L)の収量はG8Mと同等です。G8MR69H (G8LR69H) とG8MR233H (G8LR275H) は、各々G8M (G8L)のN-末端側CRDとC-末端側CRDの糖鎖結合サイトを形成するArgをHisに置換した変異体、G8MR69,233H (G8LR69,275H) は両方のCRDに変異を導入したものです。G8の場合、lactoseに対する親和性はN-末端側CRDとC-末端側CRDで大きく異なり、N-末端側CRDが高い親和性を持つ一方で、C-末端側CRDはほとんど親和性を示しません 6,7。従って、N-末端側CRDが不活性化されたR69H変異体を、lactose-agaroseを用いてアフィニティー精製することはできません。以前の論文にG8MR69Hがasialofetuin-agaroseに結合することを示すデータを載せていますが、asialofetuinの場合でもサンプル添加後のカラム洗浄を最小限に留めないと、G8MR69Hはカラムに保持されません 8。
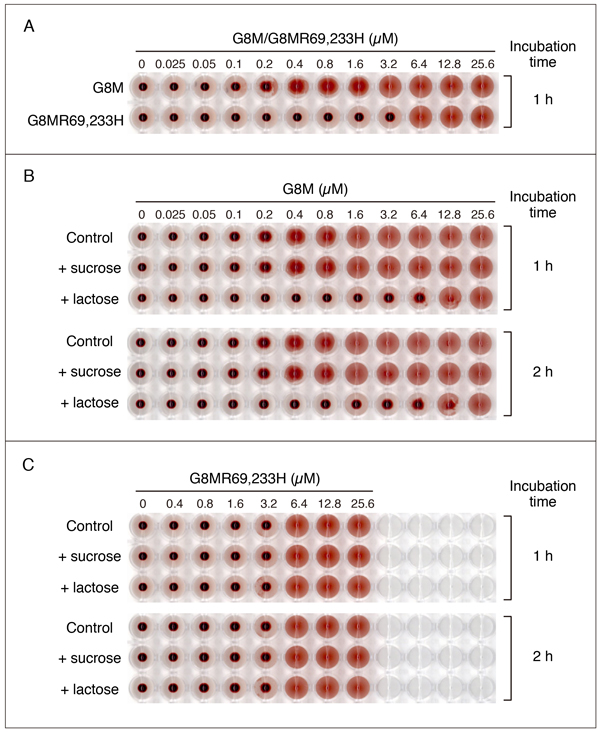
G7の項でも触れましたが、糖鎖結合サイトを形成するArgをHisに置換した変異体が、赤血球凝集活性を示す可能性があります。Fig. 8はG8MとG8MR69,233Hの赤血球凝集活性を比較した結果です。測定方法については、「補足」の項を参照して下さい。条件を揃えるため、両者ともGST-fusionとして発現させた後、GST-tagを除去した標品を用いています。ガレクチンファミリーの中で、G8Mの赤血球凝集活性は比較的低く、今回使用した条件下で活性を検出できる最小濃度は0.2 - 0.4 µMです(Fig. 8A)。G8MR69,233Hは6.4 µM以上で活性を示し、G8Mの3 - 6%程度の活性を保持している可能性があります。G8Mの作用は50 mM lactoseによって阻害されますが、高濃度(12.8, 25.6 µM)のG8Mによる赤血球凝集は阻害されにくく、インキュベーション時間を2時間に延長すると、12.8 µMでも弱い阻害効果が認められるようになります(Fig. 8B)。一方、G8MR69,233Hの場合、50 mM lactoseによる阻害は観察されず、インキュベーション時間を2時間に延長しても結果は変わりません(Fig. 8C)。G8MR69,233Hが高濃度で赤血球凝集活性を示すことは確認できましたが、これが残存するレクチン作用に基づくものかどうかは、明らかではありません。
G8LR197AはG8Lのリンカーペプチドに存在するArg197をAlaに置換した変異体です。G8Lのリンカーペプチド(G8Mには存在しない部分)にはthrombinが認識するサイト(-IAPRT-)が存在し、この部位のArg197をAlaに置換するとthrombinで切断されなくなります 9。従って、この変異を導入していないGST-G8L(pGEX-G8(L), pGEX-G8(L)R69H, pGEX-G8(L)R275H, pGEX-G8(L)R69,275Hの産物)をthrombinで処理すると、GST-tagが除去されるだけでなく、リンカーペプチドの切断によって2つのCRDが分離してしまいます。
タンデムリピート型ガレクチンに存在するリンカーペプチドは、一定の立体構造を形成しないdisorder領域(disordered region)であり(「補足」の項を参照)、様々なプロテアーゼによって容易に分解されます。組換えタンパク質の場合も、アフィニティー精製だけでは大腸菌由来のプロテアーゼの除去は完全ではありません。G8NullはG8のリンカーペプチドをほぼ除去した変異体で、プロテアーゼによる分解に対して高い耐性を示します 10。G8Null(pET-G8Null)の収量は約40 mg/L cultureです。なお、変異体発現用ベクターを作製した際に利用したNdeI siteが2つのドメインの繋ぎ目に存在しますので、このサイト(CATATG)に由来する余分な2アミノ酸残基(HM)が含まれます。G8Null, G8NT, G8CT, G8NCRD, G8CCRDの構造は以下のとおりです。アミノ酸残基番号はG8Mの番号です。
| G8Null: | N-末端側CRD*(Met1 - Ser155)+ NdeI site由来の配列(HM)+ C-末端側CRD(Arg184 - Trp317) |
| G8NT: | N-末端側CRD*(Met1 - Ser155)+ リンカーペプチドの一部(Asp156 - Thr168) |
| G8CT: | リンカーペプチドの一部(Ser171 - Leu183)+ C-末端側CRD(Arg184 - Trp317) |
| G8NCRD: | N-末端側CRD*(Met1 - Phe153) |
| G8CCRD: | C-末端側CRD(Arg184 - Trp317) |
* G8のN-末端側CRDは(Met1 - Ser155)と考えた方がより適切と思われます。
・G9
ヒトG9には、リンカーペプチドの構造のみが異なる少なくとも3種類のアイソフォーム(G9S, G9M, G9L)が存在します。リンカーペプチドは各々、33残基、45残基、77残基です。以前の論文ではC-末端側CRDをThr166 - Thr311(G9Sのアミノ酸残基番号)としましたが 10、立体構造から判断するとPro182 - Thr311の方が適切だと考えられます。G9の場合、組換えタンパク質の収量はアイソフォーム間で異なり、一般的な生化学実験に使用する量のG9Lを得るのは困難です。G9Mの収量はG9Sの30%程度であり、リンカーペプチドの分解はG9Sより速くなります。Fig. 6Bの30LE*と20LE*でバンドの濃さが異なっているのは、G4の場合と同様、30LEのタンパク質濃度が過大評価されていることが原因の1つである可能性があります。また、G9を含むいくつかのガレクチン標品で、SDS抵抗性の多量体/凝集体の形成(ゲル上端のバンド)が認められます(G13, G14/PPL13の項参照)。精製標品間での多量体/凝集体形成の差も、メインバンドの濃さに影響していると思われます。
G9MR65H, G9MR239H, G9MR65,239HはG8と同様、N-末端側CRDとC-末端側CRD(あるいは両方)の糖鎖結合サイトを形成するArgをHisに置換した変異体です。
G9NullはG9のリンカーペプチドの一部を除去した変異体ですが、上述のようにリンカーペプチドと考えられる約16アミノ酸残基と、G8の場合と同様、NdeI siteに由来する余分な2アミノ酸残基(HM)が含まれています。G9Nullはプロテアーゼによる分解に対して高い耐性を示し、また組換えタンパク質の収量も向上しています 10。G9(G9Nullを含む)は、lactose-agaroseからlactoseで溶出された時点では、ある程度高い濃度の溶液として存在しています。しかし、溶出液は直ちに濁り始め、PBSなど生理的なpHの緩衝液に対して透析すると、大部分が不溶化します。野生型より溶解性の向上したG9Nullの場合でも、PBS中で安定して存在できる濃度は400 µg/mL以下です。このため精製に際しては、通常であれば廃棄するG9濃度の低い溶出画分と高濃度の溶出画分を予め混合しておくことで、透析後の収量を上げることができます。但し、透析前に400 µg/mLまで希釈した場合でも、透析によるある程度の不溶化は避けられないため、透析前の濃度を下げすぎると、最終標品の濃度が低くなります。Table 3のG9SとG9Nullのデータはこのような方法を用いて得られたものです。G9の不溶化は、溶出液、透析外液のpHを下げることでかなりの程度防ぐことができます 11。酸性条件下での精製については「補足」の項を参照して下さい。G9NullR65D/G9NullR211DはG9MR65H/G9MR239Hに対応する変異体です。G9NullR65H/G9NullR211Hは実質的に組換えタンパク質が得られなかったため、これらの変異体を使用しています。
G9がプロテアーゼに高い感受性を示すという問題はG9Nullによって解消されましたが、生理的pHでの可溶性の問題はあまり改善していません。ssG9(highly stable and soluble form of G9)はこの問題を解決するために作製した変異体です 12。G9Nullに残っているリンカーペプチドのうち10アミノ酸残基を除去し、1つのアミノ酸置換を導入しています。この改変体は、生理的pHにおいて少なくとも2 mg/mL程度の溶液として長期間安定に保存することができます。プロテアーゼ耐性とレクチン活性はG9Nullと同等あるいはそれ以上です。ssG9は2つのCRDの間に6アミノ酸残基HPPYPM(野生型の配列はHPAYPM)が存在する構造です。余分なアミノ酸残基の挿入はありません。野生型G9 cDNAにはBamHI siteが存在しますが、ssG9(pET-ssG9)ではこの配列を変更してあります(GGATCC → GTATCC)。G9Null, ssG9, G9NT, G9CT, G9NCRD, G9CCRDの構造は以下のとおりです。アミノ酸残基番号はG9CTのみがG9M、他はG9Sの番号です。
| G9Null: | N-末端側CRD(Met1 - Gln148)+ NdeI site由来の配列(HM)+ リンカーペプチドの一部(Thr166 - Met181)+ C-末端側CRD(Pro182 - Thr311) | ssG9: | N-末端側CRD(Met1 - Gln148)+ 改変されたリンカーペプチドの一部(HPPYPM)+ C-末端側CRD(Pro182 - Thr311) | G9NT: | N-末端側CRD(Met1 - Gln148)+ リンカーペプチドの一部(Pro149 - Thr167) | G9CT: | リンカーペプチドの一部(Ile160 - Met193)+ C-末端側CRD(Pro194 - Thr323) | G9NCRD: | N-末端側CRD(Met1 - Gln148) | G9CCRD: | リンカーペプチドの一部(Thr166 - Met181)+ C-末端側CRD(Pro182 - Thr311) |
・G10
G10をlactose-agaroseを用いてアフィニティー精製することは実質的に不可能です。
精製G10標品は濃度が低いため、SDS処理する前にメタノールを使って濃縮しています(Fig. 7A, 20GE*)。濃縮方法については、「補足」の項を参照して下さい。
G4と同様、精製標品に大腸菌シャペロンが含まれていますので、GSTマニュアルで推奨されているDnaK除去方法の効果をG10で確認してみました(Fig. 7B)。GSTマニュアルではアフィニティー精製の前(菌体抽出液)にATPとインキュベートすることを推奨していますが(ATP固定化カラムの利用にも触れています)、Fig. 7Bの実験では、GST-G10がglutathione-Sepharoseに結合した状態で処理を行いました。また、thrombin処理はFig. 2の条件ではなく、GST-G10をゲルから溶出した後に行っています。GST-G10が結合したglutathione-SepharoseをATPを含む緩衝液とインキュベートすると、DnaKが遊離してきます(Fig. 7B ATP·W)。精製標品を比較すると、ATPで処理したサンプル(Fig. 7B右側のG10)では処理しないものと較べてDnaKが減少しています。GroELの含量も若干減少しているようです。
Fig. 2の2倍のスケールでStep 17まで進める。Step 18でゲルを2本のカラムにパックし(1 mL gel/column)、Step 19と同じ条件で洗浄する。一方のカラム(カラムA)から50 mM Tris-HCl (pH 8.0), 10 mM glutathioneでGST-G10を溶出する。もう1つのカラム(カラムB)に 1 mLの50 mM Tris-HCl (pH 7.5), 2 mM ATP, 10 mM MgCl2を加え(流出液は廃棄)、37°Cで10分間インキュベートする。この操作を3回繰り返し、2回目と3回目の流出液をATP洗浄画分として保存する。カラムを3 mLのTBS, 0.03% CHAPSで洗浄した後、50 mM Tris-HCl (pH 8.0), 10 mM glutathioneでGST-G10を溶出する。カラムA, Bの溶出液に30 µLのthrombin溶液(1 u/µL)を加え、25°Cで16 hインキュベートする。Fig. 2 Step 25, 26の条件で透析・遠心する。得られた上清をglutathione-Sepharoseカラム(0.5 mL gel)に流し、非吸着画分を集めて精製標品とした。各サンプル30 µLに10 µLのSDS sample buffer (4X)を加えて熱処理した後、SDS-PAGEで分析した(10 µL/lane)。
| GE: | 10 mM glutathioneによるカラム溶出画分 | GE·Th: | thrombin処理したGE | GE·Th·d: | GE·Thを透析・遠心後の上清 | G10: | 精製標品 | ATP·W: | ATP洗浄画分(カラムB) |
・G12
Table 1に記載したクローンとFig. 2の方法を用いて、可溶性のG12, LNG12, G12NT, G12CTを得ることは実質的に不可能です*。精製標品(Fig. 7C, 20GE)のメインバンドは、GroELと思われます。
G12はYang et al. 13 に記載されているクローンに対応します。Hotta et al. 14 は、Yang et al.の論文に記載されている開始コドンよりも5'上流側に、もう1つの開始コドンが存在することを報告しており、LNG12はこのクローンに対応します。LNG12はG12よりもN-末端側に22アミノ酸残基延長しています。何れの論文でも、G12タンパク質の発現確認にはウェスタンブロット法もしくは放射性アミノ酸標識を用い、精製組換えタンパク質を対象とした実験は行われていません。なお、in vitro翻訳系を利用して作製したG12([35S]Met標識)は、lactose-agaroseに弱い親和性を示します 14。G12NT, CTの構造は以下のとおりです。
| G12NT: | N-末端側CRD(Met1 - Leu161)+ リンカーペプチドの一部(Asn162 - Ser170) |
| G12CT: | リンカーペプチドの一部(Pro174 - Val188)+ C-末端側CRD(Pro189 - Ser314) |
G12の立体構造は報告されていないため(in silico analysisを除く)、CRD/リンカーペプチドの範囲は暫定的なものです。
* Maller et al. 15 は、pET-28aベクターと真核生物のタンパク質発現に適した大腸菌株(Rosseta 2(DE3)pLysS:大腸菌で使用頻度の低い7種類のコドン、AGA, AGG, CGG, GGA, AUA, CUA, CCCに対応するtRNAの発現量を高めた大腸菌株)を用いて、可溶性のマウスG12の発現・精製を報告しています。この方法で発現させたマウスG12(Hisタグ付き)は、Ni-NTA担体やlactose-Sepharoseでは精製できないため、Q-SepharoseとCM-Sepharoseを用いたイオン交換クロマトグラフィーで精製しています(収量:5 mg/L culture)。
・G13, G14/PPL13
G13, G14ともにlactose-agaroseを用いたアフィニティー精製は実質的に不可能です。
精製G14/PPL13標品は濃度が低いため、SDS処理する前にメタノールを使って濃縮しています(Fig. 7E, 20GE*)。濃縮方法については、「補足」の項を参照して下さい。なお、G14/PPL13の精製標品は多量の不純物(おそらくはGroEL)を含んでいますので、Table 2の"Protein concentration (mg/mL, A280 nm = 1.0)"、Table 3の"Purified protein yield"等は信頼できる値ではありません。
Fig. 7Dの20GE§と20GE*,§は、泳動前に熱処理を行っていないサンプルです。精製したG13標品をSDS sample buffer (1X)中で熱処理(98°C, 3 min)すると、SDS抵抗性の多量体/凝集体が形成されます(Fig. 7D, 20GE/20GE*)。同じような多量体/凝集体の形成がG9S等でも認められますが、必ずしも熱処理とは関連しません。
濃縮や透析はタンパク質を扱う際に比較的頻繁に行われる作業ですが、生物活性を損なわないことが必要な場合、煩雑で時間のかかる作業でもあります。一方、SDS-PAGE(Coomassie染色、Western blot、Lectin blotなど)のために希薄なタンパク質溶液を濃縮する際は、より簡便な方法を利用することができます。ガレクチン研究と特別な関係はありませんが、精製ガレクチン標品の濃縮に利用した方法(メタノール沈殿法とSCR吸着法)とTCA-DOC(trichloroacetic acid-deoxycholic acid)沈殿法を記載しておきます(以下メタノール法、SCR法、TCA-DOC法)。Fig. 9に3つの濃縮法の実施例を示します。
Fig. 9A, 9Bは各々、ラットの組織抽出液に上述の3種類の濃縮法を適用した結果と精製ガレクチン標品(+ BSA, bovine serum albumin)に2種類の濃縮法(メタノール法とSCR法)を適用した結果です。C(control sample)はFig. 9Aの場合7.5 µg protein/lane、Fig. 9Bの場合は1 µg protein/lane。MeOH, TCA-DOC, SCRは各々、メタノール法、TCA/DOC法、SCR法で濃縮したサンプルです。ラット組織抽出液の場合、下記の実験条件ではメタノール法が最も良い結果を示しています。SCR法では、回収率の低い、あるいはほとんど回収できないバンド(Fig. 9A, arrowheads)がいくつかあることが分かります。精製ガレクチン標品の場合もほぼ同様で、分子量がある程度大きいキメラ型ガレクチンやタンデムリピート型ガレクチンでは、メタノール法とSCR法の間で大きな違いはありませんが、SCR法ではプロトタイプ型ガレクチン(とBSA)の回収率が低下しています。SCR法は簡便で優れた濃縮法ですが、利用に際しては注意が必要です。SCRのプライミング(下記SCRの項参照)がタンパク質の回収率に与える影響については、比較実験を行っていないため、明らかではありません。Fig. 9BのG4標品は、保存に伴って生じた分解物を含んでいます。G13標品はすべて熱処理を行っていません(G13, G14/PPL13の項参照)。
・メタノール法
硫安沈殿などと同様、タンパク質沈殿法の1つです。沈殿法は、一般的には希薄なタンパク質溶液には適用できず、タンパク質濃度が1 mg/mL程度以上が目安ですが、Fig. 9のデータは、敢えて低濃度のタンパク質溶液(100 µg/mLのラット組織抽出液と20 µg/mLのガレクチン溶液)に適用した結果です。試料に対しての4倍量のメタノールを加えています(最終メタノール濃度は約80%)。
Rat tissue extract (100 µg/mL in TBS), 300 µL in a 2-mL tube
↓ + 1.2 mL of methanol, followed by mixing
↓ 30 min on ice
↓ cent., 14,000 rpm (15,000 xg at Rav), 20 min
Ppt.
↓ residual methanol was removed by an additional brief centrifugation and careful aspiration
↓ dissolved in 40 µL of SDS sample buffer (1X), and then subjected to heat treatment
SDS-PAGE
Galectin solution (20 µg/mL in PBS), 200 µL in a 1.5-mL tube
↓ + 0.8 mL of methanol, followed by mixing
↓ 30 min on ice
↓ cent., 14,000 rpm, 20 min
Ppt.
↓ residual methanol was removed by an additional brief centrifugation and careful aspiration
↓ dissolved in 40 µL of SDS sample buffer (1X), and then subjected to heat treatment
SDS-PAGE
・SCR法
SCRはヒドロキシル化されたシリカ粒子で、DNA溶液から制限酵素などを除く際に、フェノール・クロロフォルム処理に代わるものとして使われています。また、SCRが非特異的にタンパク質を吸着する性質を利用して、タンパク質溶液の濃縮にも利用することができます。SCRを用いるタンパク質の濃縮は沈殿法とは異なり、希薄なタンパク質溶液にも適用可能で、短時間で行うことができます。下記の実験例では300 µlの試料(100 µg protein/mL)あるいは200 µLの試料(20 µg protein/mL)に対して10 µlのSCR slurryを加えていますが、SCRの吸着容量内であれば、試料を増やすことができます。メタノール沈殿と同様、主としてSDS-PAGEや質量分析用の試料を調製するための方法であり、生理活性を保ったままSCRに吸着したタンパク質を回収するのは困難と思われます。界面活性剤はSCRとタンパク質の結合を阻害しますので、界面活性剤を含む溶液の場合は注意が必要です。
Otto et al.は、SCRを利用したタンパク質の濃縮と保存について詳しい検討を行っています 16。この文献に記載されている方法では、最初にSCRのプライミング(塩酸中で100°C, 6 hの熱処理)を行っていますが、下記の実験ではプライミングは行わず、市販のSCRをそのまま使用しています。なお、SCRはかなり高価な試薬です。
Rat tissue extract (100 µg/mL in TBS), 300 µL in a 1.5-mL tube
Galectin solution (20 µg/mL in PBS), 200 µL in a 1.5-mL tube
↓ + 10 µL of SCR slurry (50%[v/v] in H2O)
↓ mixed for 1 min
↓ cent., 30 sec (table top centrifuge)
Ppt.
↓ residual supernatant was removed by an additional brief centrifugation and careful aspiration
↓ suspended in 40 µL of SDS sample buffer (1X), and then subjected to heat treatment
↓ cooled on ice
↓ cent., 30 sec (table top centrifuge)
Sup.
↓
SDS-PAGE
・TCA-DOC法
この方法は、妨害物質を含む試料のタンパク質濃度をLowry法で定量するために開発されたものです17。沈殿法の1つですが、DOCを一種のキャリアとして利用することで、希薄なタンパク質溶液にも適用可能です。沈殿剤としてTCAを用いているため、メタノールでTCAを除く処理を加えています(Lowry法の試料とする場合は不要な操作です)。
Rat tissue extract (100 µg/mL in TBS), 300 µL
↓ + 300 µL of TBS
↓ + 5 µL of 2% sodium DOC*, followed by mixing
↓ 15 min at room temperature
↓ + 150 µL of 30% TCA, followed by mixing
↓ 15 min on ice
↓ cent., 14,000 rpm (15,000 xg at Rav), 20 min
Ppt.
↓ residual supernatant was removed by an additional brief centrifugation and careful aspiration
↓ + 1 mL of methanol, followed by mixing
↓ cent., 14,000 rpm, 5 min
Ppt.
↓ residual supernatant was removed by an additional brief centrifugation and careful aspiration
↓ dissolved in 40 µL of SDS sample buffer (1X), and then subjected to heat treatment
SDS-PAGE
* デオキシコール酸ナトリウムを溶解する際は、NaOH溶液でpHを8 ~ 8.5程度の弱アルカリ性に調節する。保存中に濁りを生じることがあるが、懸濁してそのまま使用可能。
赤血球凝集活性測定には様々なプロトコールが存在しますが、このノートでは下記の方法を用いました。
96-well plate, V-bottom (U-bottom)
↓ + 50 µL/well of sample (serially diluted with PBS, 10 mg/mL BSA, 0.05% NaN3)
↓ + 50 µL/well of 2% (v/v) suspension of trypsinized, glutaraldehyde-fixed rabbit erythrocytes
(diluted with PBS, 10 mg/mL BSA, 0.05% NaN3), and then mixed by pipetting several times using a multichannel pipette set at 50 µL
↓ stand for 1 h (2 h) at room temperature
The extent of hemagglutination was observed visually.
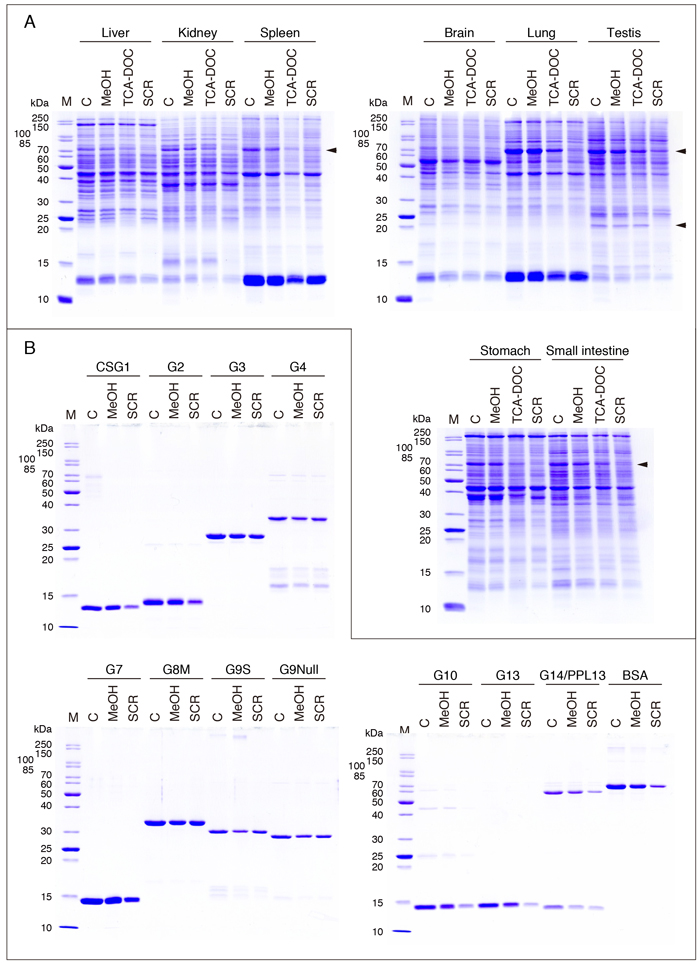
赤血球凝集活性測定では肉眼的に凝集の有無を評価するため、レクチンの性質によっては、最小有効濃度を決定するのが難しい場合があります。また、使用するプレートの形状や赤血球懸濁液の濃度も判断に影響を与えることがあります。Fig. 10にG8M, G8MR69,233H, G9Null, CSG1の例を示します。G9Nullの場合、プレートの形状(V-bottomあるいはU-bottom)は、最小有効濃度の判断に影響しませんが、G8MR69,233Hの場合は、U-bottomプレートを使用すると判断が難しくなります。また、G8MとCSG1(特にCSG1)は、濃度の上昇に伴って徐々に赤血球の凝集状態が変化するため、どちらのプレートを使っても、判断が恣意的になる可能性があります。
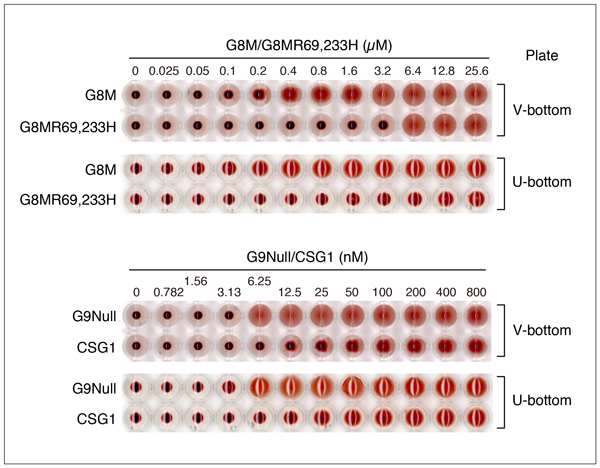
Fig. 11はガレクチン(G1 - G13)のdisorder領域予測のデータです。以前にPOODLE(Prediction Of Order and Disorder by machine LEarning)のwebサービスを利用して得られたデータです。現在このサービスは終了していますが、プログラムをダウンロードし、ローカルPC上で実行することは可能なようです(Protein Structure Prediction Active Workflow: https://togo.medals.jp/active.html)。G3のN-末端側の非レクチンドメインとタンデムリピート型ガレクチンのリンカーペプチドに相当する領域のdisorder probabilityが高くなっています。リンカーペプチドの大部分を除去したG8NullとG9Nullの場合、disorder領域と予測される部分は存在しません。他のタンデムリピート型ガレクチンとは異なり、LNG12/G12には典型的なdisorder領域は存在しないようです。ややdisorder probabilityが高くなっている領域も、リンカーペプチドと思われる領域とは一致していません。これは、G12の特殊な細胞内分布(分化した3T3-L1細胞のlipid dropletの内面に局在する)15 、と関係があるのかもしれません。プロトタイプ型に属するG7とG13にもdisorder probabilityが若干高い領域が存在しますが、理由はよく分かりません。
Fig. 1のStep 20以降を下記の方法に変更してG9Nullを発現・精製した結果を以下に示します(培養液、200 mL;20°C, 16 h)。
| 酸性条件*: | 0.708 mg/mL** x 3.85 mL |
| Fig. 1の条件*: | 0.385 mg/mL x 5.00 mL |
* 直接比較できるように、同時に2つの条件で行った実験の結果。
** G9Nullの280 nmにおける吸光係数が、pH 7.5とpH 6.0で同じと仮定した値。
精製標品のvolumeが異なっているのは、G9の項で説明したように、後者(Fig. 1の条件)では濃度の低い溶出画分を加えて透析しているためです。Fig. 1の方法ではStep 22で相当量の沈殿(不溶化したG9Null)が得られます。この沈殿から、lactoseを含む酸性の緩衝液(20 mM Na-Pi (pH 6.0), 0.15 M NaCl, 0.2 M lactose)を使ってG9Nullの回収を試みましたが、大半は可溶化されませんでした(data not shown)。
20) Wash the gel with 2 gel-bed volumes of 0.15 M NaCl.
↓
21) Elute the recombinant protein from the gel with 20 mM Na-Pi (pH 6.0), 0.15 M NaCl, 0.2 M lactose.
↓
22) Dialyze the eluate against 20 mM Na-Pi (pH 6.0), 0.15 M NaCl*.
*1st, 4 - 5 h (500 mL); 2nd, overnight (1,000 mL); 3rd, 4 - 5 h (500 mL)
↓
23) Transfer the dialyzed solution to a centrifuge tube, and then spin down the insoluble material
at 25,000 xg for 20 min.
↓
24) Sterilize the supernatant with a sterile filter (0.2 µm).
↓
25) Store the sterilized solution at 4 °C.
本稿の作成・公開にあたり、ご協力とご支援を頂きました、伊藤愛子博士(香川大学総合生命科学研究センター)、中村隆範教授(香川大学医学部)、平林淳博士(産業技術総合研究所生命工学領域)に深謝いたします。
Abbreviations
BSA: bovine serum albumin
CHAPS: 3-[(3-Cholamidopropyl)dimethylammonio]propanesulfonate
CRD: carbohydrate recognition domain
DMSO: dimethylsulfoxide
DOC: deoxycholic acid
DTT: dithiothreitol
FAC: frontal affinity chromatography
G1 - G14: galectin-1 - galectin-14
GST: glutathione S-transferase
IPTG: isopropyl β-D-1-thiogalactopyranoside
Na-Pi: sodium phosphate
PA: pyridylaminated/pyridylamination
PMSF: phenylmethanesulfonyl fluoride(別名:benzylsulfonyl fluoride等)
pNP: p-nitrophenylated/p-nitrophenylation
SCR: StrataClean Resin
TBS: Tris-buffered saline
TCA: trichloroacetic acid



