
Jin-ichi Inokuchi
Jin-ichi Inokuchi was born in Fukuoka in 1953. He received his doctorate in 1984 from the Department of Biochemistry, Faculty of Pharmaceutical Sciences, Fukuoka University under Prof. Atsuo Nagamatsu. In 1985 he moved to the Mental Health Research Institute, University of Michigan, as a postdoctoral fellow and Fulbright Senior Scientist, working under Prof. Norman Radin. During this period he contributed in particular to the development of the inhibitor of glucosylceramide synthase (PDMP). In 1992 he became manager of the Exploratory Group and head of the Glycolipid Section, Tokyo Research Institute, SEIKAGAKU CORPORATION. In 1998 he moved to Hokkaido University as an associate professor. Research projects within his group are focused on the elucidation of patho/physiological roles of membrane microdomains in life style-related diseases such as type 2 diabetes and cancer. Dr. Inokuchi is a principal investigator of CREST, Japan Science and Technology Agency, entitled “Molecular pathogenesis of type 2 diabetes via insulin signaling in membrane microdomains.” In 2006, he became a professor at the Molecular Biomembrane Institute, Tohoku Pharmaceutical University.
Membrane microdomains (lipid rafts) are now recognized as critical for proper compartmentalization of insulin signaling and much interest is focused on their role in the pathogenesis of type 2 diabetes and insulin resistance. TNFα induces insulin resistance in type 2 diabetes but its mechanism of action is not fully elucidated. Five years ago, we discovered upregulation on the expression of ganglioside GM3 and its synthase gene in adipocytes treated with TNFα as well as typical obese-diabetic animals. Subsequently, various studies have proved that the insulin metabolic signaling defect can be attributed to a loss of insulin receptors in the microdomains due to an accumulation of GM3. Our findings may explain a newfound pathological feature in life style-related diseases such as type 2 diabetes.
A pandemic of diabetes worldwide has been occurring over the last two decades. In 1985 there were 30 million diabetics while in 1995 the number was 177 million. Urgent measures are necessary, since it is estimated that 300 million diabetics will exist in 2025. In particular, type 2 diabetes constitutes 90-95% of the population with diabetes.
Insulin resistance, defined as the decreased ability of cells or tissues to respond to physiological levels of insulin, is thought to be the primary defect in the pathophysiology of type 2 diabetes (Fig. 1).1
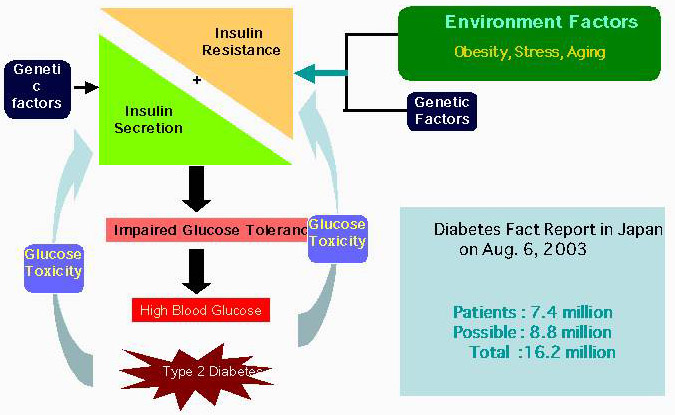
Fig. 1 Battle against insulin resistance
Since adipose tissue was found to be not only energy storage tissue but also the most active endocrine organ in the body, remarkable progress has been achieved in adipocyte biology. Especially under obese conditions, the excess accumulation of lipids in adipose tissue leads to overproduction of inflammatory cytokines such as TNFα and decreased production of adipopnectin. Such unbalanced secretion of adipocytokines strongly contributes to the development a variety pathological phenotypes in type 2 diabetes and atherosclerosis. Recently the pathophysiological role of macrophage recruitment from bone marrow into adipose tissue has been the focus of particular attention because the state of insulin resistance in white adipose tissue is induced by cytokines secreted from the invaded macrophage (Fig. 2).

Fig. 2 Pathology of insulin resistance in adipocytes
Numerous studies have implicated TNFα as having a role in insulin resistance, both in cultured adipocytes and whole-animal models.2-4 Genetically obese db/db mice, ob/ob mice and Zucker rats exhibit insulin resistance, and TNFα mRNA was expressed in their adipocytes at high levels compared to the control (lean) animals as well as in obese diabetic human subjects. When the soluble TNF receptor-IgG kimeric protein was injected into Zucker rats to absorb TNFα, the state of insulin resistance was ameliorated.4 In addition, ob/ob mice lacking TNFα gene did not develop insulin resistance.4 From these observations, considerable attention has been focused on TNFα overproduction by enlarged adipose tissues accumulating neutral fats as the cause of insulin resistance.
Adipose tissues from various species, including human, rat and mouse, contain GM3 as the most abundant type of ganglioside.5 It is known that when GM3 was added exogenously to cultured cells expressing EGF receptor6 and IR,7 inhibition of ligand-stimulated receptor autophosphorylation was observed. Moreover, there is a possiblility that endogenous GM3 may indeed regulate insulin signaling since IR exists in the membrane microdomains,8 the restricted membrane micro-compartment concentrating sphingomyelin, cholesterol and glycosphingolipids including GM3. We presented evidence for the first time that the acquisition of a state of insulin resistance in adipocytes induced by TNFα may depend on increased GM3 biosynthesis through upregulation of GM3 synthase gene expression and that GM3 may function as an inhibitor in insulin signaling during chronic exposure of adipocytes to TNFα.8 Here our recent observations demonstrating that GM3 suppressed insulin metabolic signaling by affecting the functions of microdomains are described.
Insulin elicits a wide variety of biological activities, which can be globally categorized into metabolic and mitogenic actions. (See GlycoWord GD-B03.) The binding of insulin to insulin receptor (IR) activates IR internal-tyrosine kinase activity. The activated tyrosine-phosphorylated IR was able to recruit and phosphorylate adaptor proteins such as insulin receptor substrate (IRS). The phosphorylated IRS activates PI3 kinase, resulting in the translocation of glucose transporter 4 (GLUT-4) to plasma membrane to facilitate glucose uptake. This IR-IRS-PI3 kinase signaling cascade is the representative metabolic pathway of insulin. On the other hand, the mitogenic pathway in insulin signaling initiates phosphorylation of Shc by the activated IR and then activates Ras-MAPK signaling.
When mouse adipocytes were cultured in low concentrations of TNFα which do not cause generalized suppression of adipocyte gene expression including IRS-1 and GLUT-4, interference of insulin action by TNFα occurred.9 This requires prolonged treatment (at least 72 h), unlike many acute effects of this cytokine. The slowness of the effect suggests that TNFα induces the synthesis of an inhibitor that is the actual effector. We demonstrated that the state of insulin resistance in adipocytes treated with 0.1 nM TNFα was accompanied by a progressive increase in cell surface GM3. This was reflected by increases in cellular GM3 content, GM3 synthase activity and GM3 synthase mRNA content, indicating that TNFα upregulates GM3 synthesis at the transcriptional level in cultured adipocytes.8
To elucidate whether the increased GM3 in 3T3-L1 adipocytes treated with TNFα is involved in insulin resistance, we used an inhibitor of glucosylceramide synthase, D-PDMP (See GlycoWord GL-A06),10,11 to deplete cellular glycosphingolipids derived from glucosylceramide. D-PDMP proved able to counteract TNF-induced increase of GM3 content in adipocytes and completely normalize the TNF-induced defect in tyrosine phosphorylation of IRS-1 in response to insulin stimulation.9 These findings are supported by the recent observation that knockout mouse lacking GM3 synthase exhibits enhancement of insulin signaling.12
Hotamisligil et al.13 reported that treatment of adipocytes with TNF induces an increase in the serine phosphorylation of IRS-1. This phosphorylation is an important event since immunoprecipitated IRS-1, which has been serine phosphorylated in response to TNF, is a direct inhibitor of insulin receptor tyrosine kinase activity. We have shown that TNFα induced serine phosphorylation of IRS-1 in adipocytes was completely suppressed by inhibition of GM3 biosynthesis with D-PDMP treatment, suggesting that the elevated GM3 synthesis induced by TNFα. caused the upregulation of serine phosphorylation of IRS-1.9 Since TNF-induced serine phosphorylation of IRS-1 may occur through the activation of a variety of kinases including protein kinase C, c-Jun NH2-terminal kinase, p44/42 kinase, and PI 3-kinase, it is important to identify the actual kinase(s) activated by endogenous GM3.
It was shown that adipose tissues of the obese-diabetic db/db, ob/ob, KK-Ay mice and the Zucker fa/fa rat produced significant levels of TNFα.3 Much less expression was seen in adipose tissues obtained from the lean control animals. Interestingly, these obese-diabetic animals did not show evidence of altered expression of other cytokines, such as TNFα, IL-1 or IFNγ.3,14 Thus, we were interested in measuring the expression of GM3 synthase mRNA in the epididymal fat of Zucker fa/fa rats and ob/ob mice. Northern blot analysis of GM3 synthase mRNA contents in the adipose tissues from these two typical models of insulin resistance exhibited significantly high levels compared to their lean counterparts (Fig. 3).9 We are currently analyzing the levels of GM3 expression in adipose tissues and plasma from other rodent models as well as from humans with type 2 diabetes.
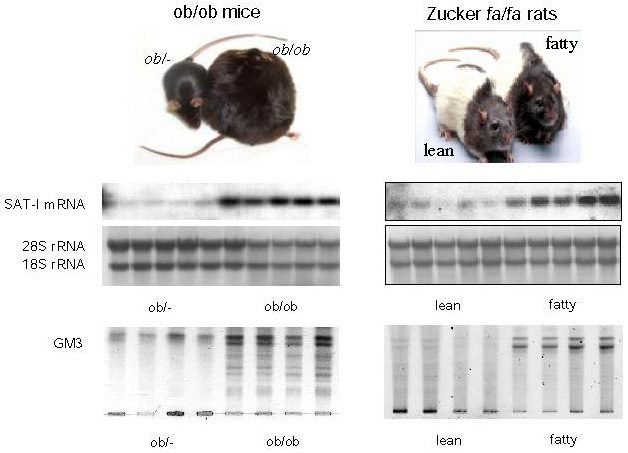
Fig. 3 Significant increase of the SAT-1 gene and GM3 in obese diabetes models
Cellular membranes contain sub-domains called detergent-resistant microdomains (DRMs) because they are detergent-insoluble and highly enriched in cholesterol and glycosphingolipids (GSLs), but lacking in phospholipids.15,16 Within the past decade, data have emerged from many laboratories implicating these lipid microdomains as critical for proper compartmentalization of insulin signaling in adipocytes (reviewed in references 17 and 18. Caveolae are a subset of membrane microdomains particularly abundant in adipocytes.19 Critical dependence of the insulin metabolic signal transduction on caveolae/microdomains in adipocytes has been demonstrated.20 Disruption of microdomains by cholesterol extraction with β-cyclodextrin resulted in progressive inhibition of tyrosine phosphorylation of IRS-1 and activation of glucose transport in response to insulin although autophosphorylation of IR and activation of MAP kinase were not impaired.20 Similarities between these cell culture results and the findings in many cases of clinical insulin resistance1 suggest a potential role for microdomains in the pathogenesis of this disorder.
Couet et al. demonstrated the presence of a caveolin binding motif (φXXXX φXXφ) in the β subunit of IRs that could bind to the scaffold domain of caveolin.21 Moreover, mutation of this motif resulted in the inhibition of insulin signaling.22 Indeed, mutations of the IR β subunit have been found in type 2 diabetes patients.23 Recently, Lisanti's laboratory reported that caveolin-1-null mice developed insulin resistance when placed on a high fat diet.24 Interestingly, insulin signaling, as measured by IR phosphorylation and its downstream targets, was selectively decreased in the adipocytes of these animals while signaling in both muscle and liver cells was normal. This signaling defect was attributed to a 90% decrease in IR protein content in the adipocytes, with no changes in mRNA levels, indicating that caveolin-1 serves to stabilize the IR protein.18,24 These studies clearly indicate the critical importance of the interaction between caveolin and IR in executing successful insulin signaling in adipocytes.
Although the direct interaction between caveolin-1 and IR has been shown as described above, studies of the presence of IRs in DRMs/caveolae have provided conflicting data.25-29 Saltiel and colleagues found that insulin stimulation of 3T3-L1 adipocytes was associated with tyrosine phosphorylation of caveolin-1.30 However, since only trace levels of IR were recovered in the caveolae microdomains in assays with a buffer of 1% Triton X-100, they speculated on the presence of intermediate molecule(s) bridging IR and caveolin.28 Gustavsson et al. also observed the dissociation of IRs from caveolin-containing DRMs after treatments of 0.3 and 0.1% Triton X-100.26 It has been reported that a comparison of protein and lipid contents of DRMs prepared with a variety of detergents indicated considerable differences in their ability to selectively solubilize membrane proteins and to enrich sphingolipids and cholesterol over glycerophospholipids and that Triton was the most reliable detergent.31 Therefore, we performed a flotation assay with a wide range of Triton X-100 concentrations to identify the protein of interest which might weakly associate with DRMs. In an assay system containing less than 0.08% Triton X-100, we were able to show that in normal adipocytes IRs can localize to DRMs. Thus, by employing low detergent concentrations we were able to demonstrate, for the first time, the presence of IR in DRMs.32 As summarized in Table 1, there are many evidence demonstrating that the localization of IRs in caveolae microdomains are essential for successful metabolic signaling of insulin.
Table 1 Localization of insulin receptor in caveolae microdomains is essential for metabolic signaling of insulin

In a state of insulin resistance induced in adipocytes by TNFα, we presented evidence that the transformation to a resistant state may depend on increased ganglioside GM3 biosynthesis following upregulated GM3 synthase gene expression. Additionally, GM3 may function as an inhibitor of insulin signaling during chronic exposure to TNFα.9 These findings are further supported by a recent report that mice lacking GM3 synthase exhibit enhanced insulin signaling.12 Since GSLs, including GM3, are important components of DRMs/caveolae, we have pursued the possibility that increased GM3 levels in DRMs confer insulin resistance upon TNFα-treated adipocytes. We examined the effect of TNFα on the composition and function of DRMs in adipocytes and demonstrated that increased GM3 levels result in the elimination of insulin receptors (IRs) from the DRMs while raft marker proteins such as caveolin and flotillin remain in the DRMs.32 Although the localization of IRs to DRMs may be maintained by the association with caveolin-1 as mentioned above, the excess accumulation of GM3 in the DRMs may weaken IR-caveolin interaction. Indeed, IR, but not caveolin-1, was co-immunoprecipitated with anti-GM3 antibody (unpublished observation). Further work is in progress to elucidate the mechanisms for the interactions of the ganglioside GM3, IR and caveolin in the microdomains.
The current view of microdomains in the state of insulin resistance is depicted in Fig. 4. We propose the following working hypothesis: "Life style-related diseases, such as type 2 diabetes, are membrane microdomain disorders caused by aberrant expression of glycosphingolipids." Based on this premise, we will explore new therapeutic strategies for type 2 diabetes insulin resistance, such as microdomain orthosignaling therapy.
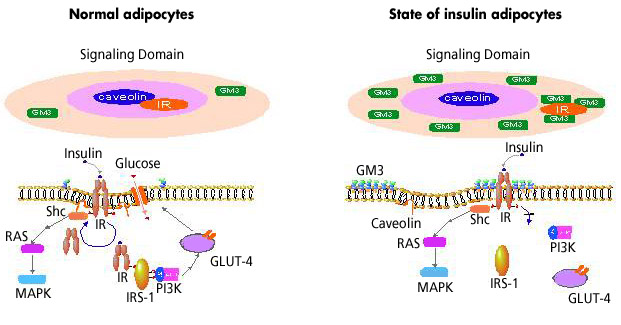
Fig. 4 Insulin resistance as a microdomain disorder



