

|
|
Type2 Diabetes and Ganglioside GM3 |
|||||||||||||||||||||||
 |
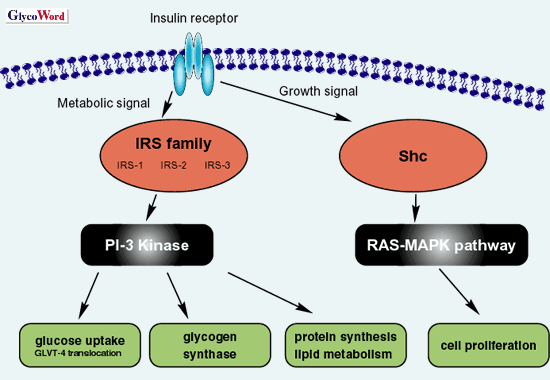
TNF Insulin elicits a wide variety of biological activities, which can be globally categorized into metabolic and mitogenic actions (Fig. 1). The binding of insulin to insulin receptor (IR) activates IR internal-tyrosine kinase activity. The activated tyrosine-phosphorylated IR was able to recruit and phosphorylate the adaptor proteins such as insulin receptor substrate (IRS). The phosphorylated IRS activates PI3 kinase, resulting in the translocation of glucose transporter 4 (GLUT-4) to plasma membrane to facilitate glucose uptake. This IR-IRS-PI3 kinase signaling cascade is the representative metabolic pathway of insulin (Fig. 1). On the other hand, the mitogenic pathway in insulin signaling initiates phosphorylation of Shc by the activated IR and then activates Ras-MAPK signaling (Fig. 1). |
||||||||||||||||||||||
 |
|||||||||||||||||||||||
|
|||||||||||||||||||||||
| In 1970, Berson and Yalow defined insulin resistance as
"Decreased ability of cells or tissues to respond to physiological
levels of insulin.” In the developed countries, pathological significance
of insulin resistance is highly recognized as the cause of lifecycle -
related diseases including hyperlipidemia, hypertension, hyperinsulinemia,
and more significantly, obesity and type 2 diabetes. In the state of insulin
resistance, the metabolic signals of insulin in liver, muscle and adipose
tissues were selectively impaired. In fact, knockout mice lacking IRS-1
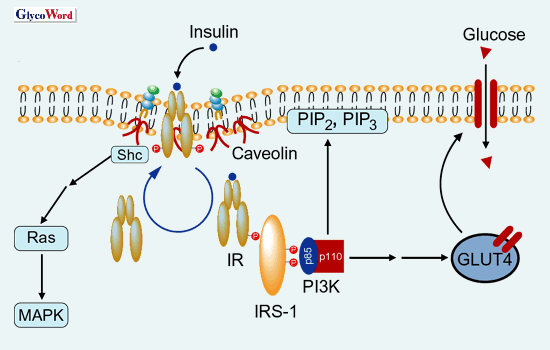
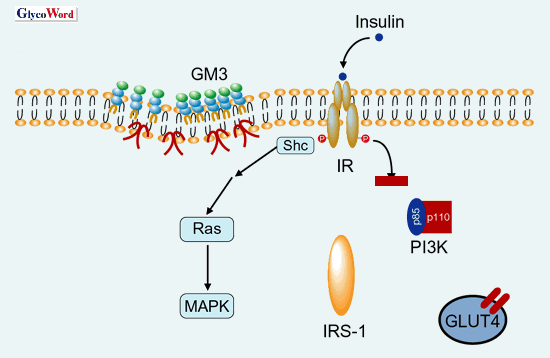
exhibited insulin resistance with hyperinsulinemia 1). Adipose tissues from various species, including human, rat and mouse, contain GM3 as the most abundant type of ganglioside. It is known that, when GM3 is added exogenously to cultured cells expressing EGF receptor 2) and IR 3), the inhibition of ligand-stimulated receptor autophosphorylation is observed. Moreover, there is a possiblility that endogenous GM3 may indeed regulate insulin signaling since IR exists in the membrane microdomains 4), the restricted membrane micro-compartment concentrating sphingomyelin, cholesterol and glycosphingolipids including GM3. We present evidence for the first time that the acquisition of a state of insulin resistance in adipocytes induced by TNF Insulin resistance in type 2 diabetes and TNF Genetically obese db/db mice, ob/ob mice and Zucker rats exhibit insulin resistance, and TNF Ganglioside GM3 as inducer of insulin resistance 5) When mouse 3T3-L1 adipocytes were cultured in low concentrations of TNF To elucidate whether the increased GM3 in 3T3-L1 adipocytes treated with TNF Regulatotry roles of microdomains in insulin signaling Insulin-mediated internalization of IR into endosomes from plasma membranes and the intracellular movement of IRS-I to endosomes were significantly suppressed in 3T3-L1 adipocytes treated with TNF Further work is in progress in our laboratory to pursue the functional and structural changes of microdomains in the state of insulin resistance and type 2 diabetes to elucidate the fundamental roles of GM3 employing bioinformatics including proteome, metabolome and structural biology. |
|||||||||||||||||||||||
 |
|||||||||||||||||||||||
|
|||||||||||||||||||||||
 |
|||||||||||||||||||||||
|
|||||||||||||||||||||||
| Jin-ichi Inokuchi (Hokkaido University) | |||||||||||||||||||||||
|
|||||||||||||||||||||||
| Apr. 1, 2003 | |||||||||||||||||||||||
|
|
|||||||||||||||||||||||
|
|||||||||||||||||||||||

