
Tamao Endo
Dr. Endo graduated from the Faculty of Pharmaceutical Sciences, the University of Tokyo, in 1977 and obtained his Ph.D. at the same institution in 1982 under the guidance of Prof. Shoshichi Nojima. He was a postdoctoral fellow at the Baylor College of Medicine under Prof. Donald M. Marcus, and a research associate in the Institute of Medical Science, the University of Tokyo, under Prof. Akira Kobata. Since 1994, he has been the head of the Department of Glycobiology, Tokyo Metropolitan Institute of Gerontology (TMIG). TMIG was reestablished as a new foundation in 2002 and Dr. Endo is the head of the Glycobiology Research Group, Tokyo Metropolitan Institute of Gerontology, Foundation for Research on Aging and Promotion of Human Welfare. His current research interests include glycobiology on aging, dementia, and development.
Muscular dystrophies are genetic diseases that cause progressive muscle weakness and wasting1. The causative genes of several muscular dystrophies have been identified in the past fifteen years. The best known is the one described by Duchenne that results from mutations in the gene encoding a protein called dystrophin. Another subclass is congenital muscular dystrophies, where muscle weakness is apparent at birth or shortly afterwards. Recent data suggest that the aberrant protein glycosylation of a specific glycoprotein, α-dystroglycan, is the primary cause of some forms of congenital muscular dystrophy. This article reviews new insights into the glycobiology of muscular dystrophy and neuronal migration disorder.
Dystroglycan is encoded by a single gene and cleaved into two proteins, α-dystroglycan and β-dystroglycan, by posttranslational processing. In skeletal muscle, dystroglycan is a component of the dystrophin-glycoprotein complex (DGC)(Fig. 1). α-Dystroglycan is an extracellular peripheral membrane glycoprotein anchored to the cell membrane by binding to a transmembrane glycoprotein, β-dystroglycan. The α-dystroglycan-β-dystroglycan complex is widely expressed in a broad array of tissues and is thought to stabilize the plasma membrane by acting as an axis through which the extracellular matrix is tightly linked to cytoskeleton. This is because α-dystroglycan strongly binds to laminin in the extracellular matrix, and the cytoplasmic domain of β-dystroglycan interacts with dystrophin, which in turn binds to the actin cytoskeleton2.
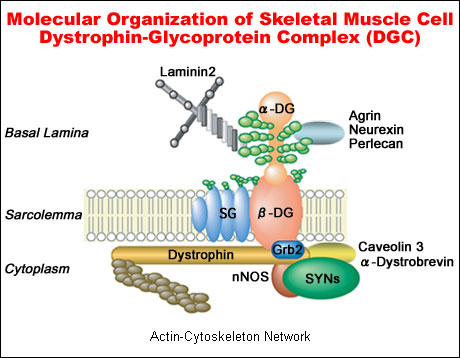
Fig. 1 Dystrophin-glycoprotein complex (DGC) and linkage between laminin-2 in the extracellular matrix and actin in the subsarcolemmal cytoskeleton.
α-Dystroglycan is a key component of the DGC and is modified by O-mannosyl glycan and binds laminin 2 via its glycan. α-Dystroglycan is also known to bind to other extracellular matrix proteins containing LamG domains, such as neurexin, agrin and perlecan. On the other hand, inside the cell, β-dystroglycan is known to bind to several components directly or indirectly. DGC, Dystrophin-glycoprotein complex; αDG, α-dystroglycan; βDG, β-dystroglycan; SG, sarcoglycan; nNOS, neural NO synthetase; and SYNs, syntrophin complex.
α-Dystroglycan is heavily glycosylated and its sugars have a role in binding to laminin, neurexin and agrin3, 4. We previously demonstrated that the sialic acid residues, which are probably attached to O-linked oligosaccharides, were essential for this binding. Further, we have elucidated the structure of O-linked oligosaccharides of bovine peripheral nerve α-dystroglycan as a novel O-mannosyl glycan, Siaα2-3Galβ1-4GlcNAcβ1-2Man3. In another study, we found the same O-mannosyl glycan in rabbit skeletal muscle α-dystroglycan4. We also found that a sialyl O-mannosyl glycan, Siaα2-3Galβ1-4GlcNAcβ1-2Man, is a laminin-binding ligand of α-dystroglycan3. After our report on the sialyl O-mannosyl glycan, a series of mammalian type O-mannosyl glycans with heterogeneous mannose-branching (2-substituted and 2,6-disubstituted mannoses) and peripheral structures [Siaα2-3Galβ1-4GlcNAcβ1-2Man, Galβ1-4GlcNAcβ1-2Man, Galβ1-4(Fucα1-3)GlcNAcβ1-2Man, HSO3-3GlcAβ1-3Galβ1-4GlcNAcβ1-2Man, Siaα2-3Galβ1-4GlcNAcβ1-2 (Siaα2-3Galβ1-4GlcNAcβ1-6) Man] have been found. Thus, mammalian O-mannosylation is a rare type of protein modification that is observed in a limited number of glycoproteins of brain, nerve and skeletal muscle4. On the other hand, O-mannosylation is known as a yeast-type modification, and oligomannose-type O-mannosylated glycoproteins are abundant in the yeast cell wall. Future studies are needed to clarify the distribution of such O-mannosyl glycans in various mammalian tissues.
Analysis of the biosynthetic pathway of the O-mannosyl glycans in mammals is important for elucidating not only the regulation of expression but also the biological functions of these glycans. The identification and characterization of the enzymes involved in the biosynthesis of mammalian type O-mannosyl glycans will be an important step forward in elucidating these glycans. A key difference between mammalian and yeast-type O-mannosyl glycans is that those in mammals have the GlcNAcβ1-2Man linkage as described above. This linkage is assumed to be catalyzed by a glycosyltransferase, UDP-N-acetylglucosamine: protein O-mannose β1,2-N-acetylglucosaminyltransferase (POMGnT1). POMGnT1 catalyzes the transfer of N-acetylglucosamine from UDP-GlcNAc to O-mannosyl glycoproteins. After we succeeded in developing an enzyme assay for POMGnT1, its activity was found in brain homogenates of several mammals5. It should be noted that GlcNAcβ1-2Man linkages are also found in N-glycans, where they are catalyzed by two enzymes, UDP-N-acetylglucosamine: α-3-D-mannoside β-1,2-N-acetylglucosaminyltransferase I (GnT-I) and UDP-N-acetylglucosamine: α-6-D-mannoside β-1,2-N-acetylglucosaminyltransferase II (GnT-II). However, we found that recombinant GnT-I and GnT-II had no ability to catalyze the GlcNAcβ1-2Man linkage in O-mannosyl glycans, suggesting that a new enzyme must be responsible for the formation of this linkage. Thus, we cloned the human POMGnT1 gene on the basis of human cDNA sequences homologous to human GnT-I (6). The nucleotide sequence indicated that POMGnT1 is a 660 amino acid protein with a calculated molecular mass of 71.5 KDa. A hydrophobicity analysis and secondary structure prediction of the amino acid sequence suggested that human POMGnT1 is a type II membrane protein. This topology was similar to the topologies of other Golgi glycosyltransferases.
The human POMGnT1 gene exists at 1p33, and we find it to be the POMGnT1 gene is located within the small candidate interval for muscle-eye-brain disease [MEB: OMIM 253280, OMIM = Online Mendelian Inheritance in Man (http://www.omim.org/)]. MEB is an autosomal recessive disorder characterized by congenital muscular dystrophy, ocular abnormalities and brain malformation (type II lissencephaly)7. Patients with MEB show severe cerebral and ocular anomalies, but some patients reach adulthood. MEB has been observed mainly in Finland. Since defects of DGC cause muscular dystrophies1, 2 and O-mannosyl type glycan is required for the laminin binding of α-dystroglycan in DGC3, it is possible that mutations in the POMGnT1 gene are related to MEB.
To test this hypothesis, we screened the entire coding region and the exon/intron flanking sequences of the POMGnT1 gene for mutations in patients with MEB. We identified six independent disease-causing mutations in these patients6. Later we found seven additional mutations and so far we have found 13 disease-causing mutations in patients with MEB8. We have not detected these 13 substitutions in any of 300 normal chromosomes, indicating that these mutations are pathogenic and that the POMGnT1 gene is responsible for MEB. To confirm that the mutations observed in patients with MEB are responsible for the defects in the synthesis of O-mannosyl glycan, we expressed the all mutant proteins and found a loss of enzymatic activity6, 9. Taking these findings together, we conclude that MEB is inherited in a loss-of-function manner of the POMGnT1 gene. If POMGnT1 does not work, no peripheral structure (Siaα2-3Galβ1-4GlcNAc, Galβ1-4(Fucα1-3)GlcNAc, and HSO3-3GlcAβ1-3Galβ1-4GlcNAc) can be formed on O-mannosyl glycan. Because these structures are known to play an important role in adhesive processes, a defect of O-mannosyl glycan may severely affect cell migration and cell adhesion. Additionally, we found a selective deficiency of α-dystroglycan in MEB patients10. This finding suggests that α-dystroglycan is a potential target of POMGnT1 and that hypoglycosylation of α-dystroglycan may play a critical role in the pathomechanism of MEB. MEB muscle and brain phenotypes can be explained by a loss of function of α-dystroglycan due to abnormal O-mannosylation.
Recent investigations have revealed that some muscular dystrophies may be caused by abnormal glycosylation of α-dystroglycan, e.g., Fukuyama-type congenital muscular dystrophy (FCMD: OMIM 253800), congenital muscular dystrophy type 1C (MDC1C: OMIM 606612), Walker-Warburg syndrome (WWS: OMIM 236670) and the myodystrophy (myd) mouse, although the details of the defects are still unclear (Table 1). Identification of these defects provides new directions for unraveling a glycopathomechanism for muscular dystrophy.
Table. 1 Possible muscular dystrophies caused by abnormal glycosylation of α-dystroglycan

Like MEB, FCMD and WWS are autosomal recessive disorders that are characterized by congenital muscular dystrophy, lissencephaly and eye anomalies11, 12. FCMD is a relatively common autosomal recessive disorder in the Japanese population11. It is the second most common form of childhood muscular dystrophy in Japan after Duchenne muscular dystrophy. Based on the averaged incidence of 3/100,000, one in ~90 persons could be a heterozygous carrier in Japan. Kobayashi et al. previously identified on chromosome 9q31 the gene responsible for FCMD, which encodes a novel 461-amino-acid protein named fukutin13. Fukutin, a protein of unknown function, has an N-terminal hydrophobic region suggesting a signal sequence or a transmembrane domain. A sequence analysis predicts it to be an enzyme that modifies cell-surface glycoproteins or glycolipids. Recently, Takeda et al. generated chimeric mice using embryonic stem cells targeted for the fukutin gene14. These mice developed severe muscular dystrophy, with the selective deficiency of α-dystroglycan and its laminin-binding activity. In addition, these mice showed the occurrence of central nervous and ocular abnormalities. Taken together, fukutin is necessary for the maintenance of muscle integrity, cortical histogenesis and normal ocular development, and a functional linkage between fukutin and α-dystroglycan is suggested.
WWS is another extreme of congenital muscular dystrophy, which shows the most severe brain malformation characterized by type II lissencephaly and eye involvement. Patients with WWS are severely affected from birth and it is usually fatal within the first year of life12. WWS has a worldwide distribution. Recently, 20% of WWS patients (6 of 30 unrelated WWS cases) have been found to have mutations in protein O-mannosyltransferase 1 (POMT1), a putative O-mannosyltransferase that catalyzes the transfer of mannose to a serine or threonine residue on the basis of homology with the seven yeast O-mannosyltransferases15. POMT1 is highly expressed in fetal brain, testis and skeletal muscle, tissues affected in WWS. It is noteworthy that none of the 30 cases studied had mutations in another homologue, POMT2. This suggests that other as yet unidentified genes are responsible for this syndrome. However, it is unclear whether the POMT1 and POMT2 proteins actually catalyze the O-mannosylation. Detection of protein O-mannosyltransferase activity of POMTs in vertebrates has not been successful. As with MEB and FCMD, a highly glycosylated α-dystroglycan was selectively deficient in skeletal muscle from WWS patients, while β-dytroglycan and laminin α2 were still expressed3, 10, 15, 16. It is noteworthy that the rt mutant in Drosophila showed defects of myogenesis caused by a homologue of POMT1 (17). Although the rt gene product is not known to initiate the biosynthesis of O-mannosyl glycan, O-mannosylation may be essential for muscular development in both vertebrates and invertebrates.
Additionally, defective glycosylation of α-dystroglycan has been implicated in congenital muscular dystrophy type 1C (MDC1C), which is caused by a homologue of fukutin (fukutin-related protein, FKRP). MDC1C is characterized by severe muscle weakness and degeneration, and cardiomyopathy. Mental retardation and cerebellar cysts have been observed in some forms. Allelic mutations in the FKRP gene also cause a milder and common form of limb-girdle muscular dystrophy2I (LGMD2I: OMIM 607155), a form frequently associated with cardiomyopathy and a variant onset ranging from adolescence to adulthood18. Patients with mutations in the FKRP gene have reduced expression of α-dystroglycan invariably, broadly correlating with disease severity, and western blot analysis shows an apparent loss of higher molecular weight forms of α-dystroglycan. Although the function of FKRP is unknown, it is suggested that FKRP is involved in the glycosylation of α-dystroglycan as a glycosyltransferase or a kind of modulator. Because FKRP and fukutin are thought to be Golgi-resident proteins19, it is possible that defects of these proteins cause abnormal processing of α-dystroglycan.
Finally, the gene large, which is mutated in the myodystrophy (myd) mouse, encodes a putative glycosyltransferase20. However, its biochemical activity has not yet been confirmed. The causative mutation in myd was identified as a deletion of exons 5-7 of the large gene. This deletion results in a frameshift in the corresponding mRNA, leading to a premature termination codon. The myd mouse showed a progressive muscular dystrophy, ocular defects, and a central nervous system phenotype characterized by abnormal neuronal migration in cerebral cortex, cerebellum and hippocampus, and disruption of the basal lamina21, 22. Hypoglycosylation of α-dystroglycan in myd muscle and brain was observed similar to the case of MEB and FCMD patients.
In summary, hypoglycosylated α-dystroglycan in the muscle membrane of MEB, FCMD and the myd mouse has greatly reduced affinities for laminin, neurexin and agrin22. Taken together, these findings suggest that defective glycosylation of α-dystroglycan due to the primary genetic defects of glycosyltransferases may be the common denominator causing muscle cell degeneration and abnormal brain structure in MEB, WWS, FCMD, MDC1C and the myd mouse. In other words, interference in glycosylation of α-dystroglycan may lead to a combination of muscle and brain phenotypes of these diseases. However, the substrates of these enzymes, with the exception of POMGnT1, are largely unknown (Fig. 2). Identification and characterization of each enzyme will help reveal the molecular pathomechanisms of congenital muscular dystrophies with brain malformation.
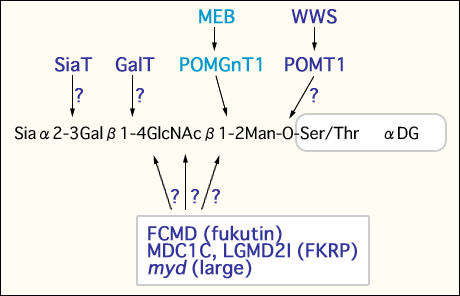
Fig. 2 Possible defects of O-mannosylglycosylation of α-dystroglycan in muscular dystrophy.
Mutations in POMGnT1, POMT1, fukutin, FKRP and large cause defects in the glycosylation of α-dystroglycan resulting in muscular dystrophy. The substrates of these putative enzymes, with the exception of POMGnT1, are largely unknown. POMT1 is thought to initiate the biosynthesis of O-mannosyl glycan. It is unclear whether other as yet uncharacterized forms of muscular dystrophy may be caused by galactosyltransferase and sialyltransferase.
Defects in glycosylation pathways in human disease were initially identified in 1980, and since then over ten congenital disorders of glycosylation (CDG) have been identified to date. They almost invariably affect N-glycan assembly (type I) or processing (type II) as reviewed in this series by Dr. Freeze23. As reviewed here, however, some forms of muscular dystrophy may be associated with mutations in genes encoding for glycosyltransferases not involved in N-glycan biosynthesis. Elucidation of the intrinsic characters of these gene products will help improve out understanding of the pathomechanisms of these complicated diseases. Hypoglycosylation of α-dystroglycan, a peripheral membrane glycoprotein expressed in muscle, brain and other tissues, is a common feature in these diseases. α-Dystroglycan may be a potential target of new glycotherapeutics strategy for muscular dystrophy as well as neuronal migration disorder in future studies.



