
氏名:Hudson H. Freeze
Freeze博士は1968年Indiana大学の微生物学教室を卒業したが、そこでT. D. Brock氏と共にTaqポリメラーゼの原料となるThermus Acuaticusの元株を発見した。その後ラホイヤにあるUCSDへ移り、1976年生物学で博士号を授与された。USCDの医学部および神経科学部でポスドクの仕事をし、1983年、医学部で兼任教官となった。ここでは、変形粘菌Dictyosteliumのリソゾーム酵素について研究し、そのオリゴ糖構造がヒトのリソゾーム酵素のものと似ていることを見出した。1988年、ラホイヤがん研究センターと呼ばれたBurnham Instituteに移り、1994年教授に指名された。博士の先天性糖鎖形成異常症(CDG)に関する興味は、未知の欠損を持つCDG患者の線維芽細胞から得られたオリゴ糖と、糖鎖形成の突然変異を起こしたDictyosteliumからの糖鎖が似ていたことに始まった。ここ数年Freeze博士は数種の新しいタイプのCDGを発見し、CDG患者に対し検査を果敢に行うよう臨床医に奨励している。2000年からはBurnham Instituteの糖鎖生物学プログラムのディレクターであり、CDGの新たな患者とタイプの同定を続けている。
本質的に全ての分泌タンパク質と細胞表面のタンパク質には糖鎖がついている。糖鎖の機能はその複雑な構造と同様に変化に富んでいるが、とりわけ細胞−細胞間のコミュニケーション、細胞内シグナリング、タンパク質の折りたたみ、細胞内でのタンパク質のターゲティングとホルモンレベルの制御がある。文字通り何千もの糖鎖構造がある。直鎖状のDNAやタンパク質と違って、糖鎖は分岐している。多くの糖鎖が修飾を担い、その上に次の糖鎖修飾が積み重なる。その一つずつで分子構造は著しく変わり、より高度に精緻な構造を作ることが出来る。複雑なために精密な構造解析が難しくなり、怖じけてしまう。糖鎖機能と糖鎖形成の研究はやりがいの有ることである。大学院生の生物医学プログラムの一部になっていることはほとんどなく、必然的に医学教育には含まれていない。臨床医がこのことに気づくことは滅多にない。臨床をやっている糖鎖生物学者はごく希であり、研修中の臨床医もこの分野に触れることが無いからである。しかしながら今や、糖鎖形成の変化が病気を引き起こすことがわかってきた。この状況は変わりつつある。
N-結合型(アスパラギン結合型)糖鎖形成の概略
全てのN-結合型糖鎖は共通のドリコールピロリン酸 (Dol-PP)-結合14糖オリゴ糖に由来する(LLO)。このものは3個のグルコース (Glc)、9個のマンノース (Man)と2個のN-アセチルグルコサミン(GlcNAc) 残基、Glc3Man9GlcNAc2からなり、脂質のキャリアからタンパク質へ固まりとして(en bloc)移される。個々の糖鎖は、最低13種の酵素により特定の順番で付加され、LLOが伸びる。その後オリゴ糖は小胞体膜に局在するオリゴ糖転移酵素複合体によって、新しく合成されたポリペプチド鎖に転移する。転移後糖鎖はプロセッシングされる。特定のグリコシダーゼがGlc3Man9GlcNAc2鎖に作用し、グルコース全部と幾つかのマンノースが小胞体中で除かれる。しばしば、さらにもう1つのマンノースがゴルジ中で取り除かれ、次いでN-アセチルグルコサミン、ガラクトース、そして最後にシアル酸(Sia)から成る2-4糖の枝が付加され、複合型の糖鎖が形成される。
先天性糖鎖形成異常症(CDG)1,2
先天性糖鎖形成異常症(CDG)は、小胞体の内腔の新しく合成されたタンパク質上のアスパラギン残基へつくN-結合型糖鎖が欠損することで生じる(それらの幾つかを図1に示した)。
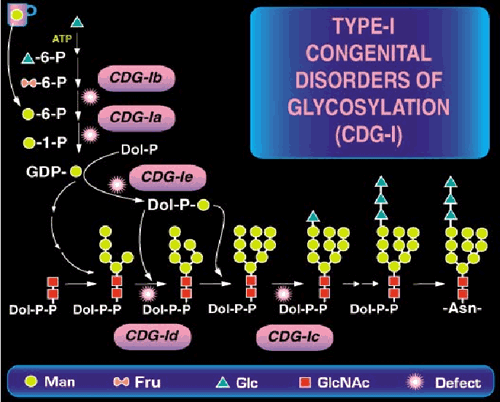
図 1 N-結合オリゴ糖生合成経路と既知欠損のある位置
引き続いて起きるオリゴ糖のプロセッシング中で、これらの糖鎖の多くは切り取られ、再度、種々の分岐型を持つ他の糖鎖が付くことで伸長する。この知見は最先端のことなので、遺伝性疾患を持つ子供の診断時、糖鎖の形成異常を考える医者はめったにいない。ちょうど5年前、我々はたった2つCDGの原因を見つけたが、今ではN-結合型糖鎖合成経路について14のCDGが知られている。図2には、ここ数年で新しい欠損がどんどん見つかってきていることを示した。
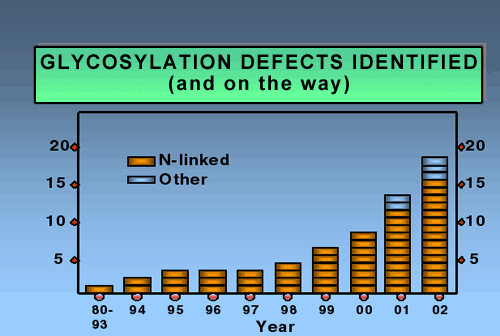
図 2 同定された糖鎖形成欠損数
N-型糖鎖形成に必要な50以上の遺伝子のいずれかが欠損するとCDGが発症し、これらの多くは簡単な血液検査で検出可能と推察される。現在世界中で約300-400例のCDG患者がいるが、信頼筋の予測では、この数値は実際患者数のほんの数%らしい。CDG全グループの診断はほとんど手つかずの状態であり、我々が見ているのは「氷山の一角」と思った方がよい。
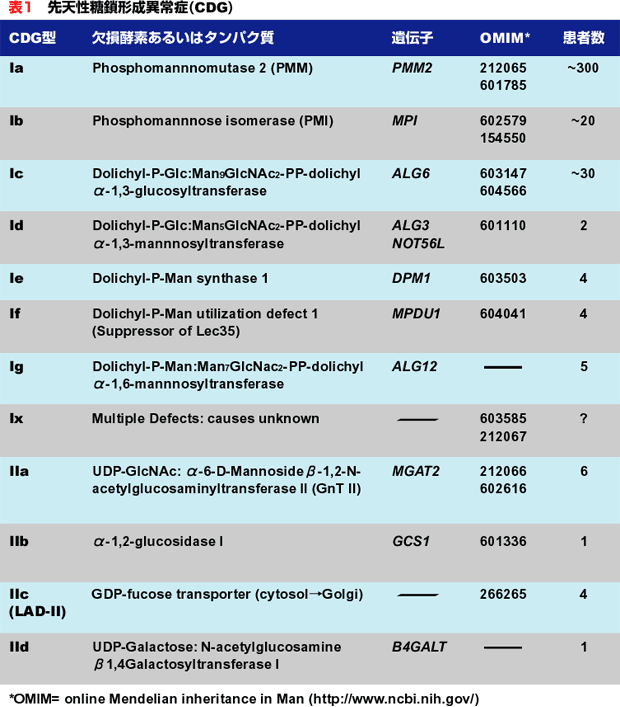
表 1に疾患タイプ、その欠損酵素、OMIMリンク、患者既知数を示した。患者の多くは当初誤診された。この状況は著しく改善されつつあるが、疾患の内2つ(CDG-IbとCDG-IIc)は単純な単糖を投与することで治療可能であるからことから、重要なことである。
CDGの生化学的概略
CDGは常染色体性劣性遺伝子疾患である。非常に希な疾患で、CDG-Ia (OMIM 212066)はPMM2遺伝子の突然変異で起きる。この遺伝子はマンノース-6-リン酸からマンノース-1-リン酸への変換で使われるホスホマンノムターゼをコードしている。変異はGDP-Manプールを減少させ完全な糖鎖形成を行うには能力不足のLLOを産生する。これらの患者の数人はミトコンドリア病と誤解されていた。CDG-Ib (OMIM 602579)はホスホマンノースイソメラーゼ (PMI) (フコース-6-P→マンノース-6-P)をコードしているMPI遺伝子の変異によって起きる。CDG-IbとCDG-Ia患者の臨床的特徴は全く異なる。CDG-Ic (OMIM 603147)はALG6の変異によって起きるが、このものは最初にグルコースが未熟なLLO前駆体に付加される時に使われるα-1,3グルコシルトランスフェラーゼをコードしている。II型のCDGは、LLO合成あるいはタンパク質への転移の段階ではなく、タンパク質−結合糖鎖のプロセッシングに影響を及ぼすものと定義されている。
CDGのラボ診断
血清トランスフェリン (Tf)の糖鎖形成はCDGの生化学的診断に使われる。異常Tfが等電点電気泳動あるいはエレクトロスプレーイオン化法(ESI-MS)で検出できる。これらの分析法はどちらも、どんな遺伝子が欠損しているかを教えてはくれないが、欠損が有ることの手がかりになる。
CDGに共通な臨床的特徴
それぞれのCDG型の最も共通する臨床的特徴を表2に示した。かなり臨床的に異なっている。中等度から重度の精神運動性の発達遅滞や弛緩が、Ib型以外の全ての患者に一定して見られる。他の神経学的所見としては運動失調 (IaとIc)、発作と脳卒中様のエピソードがある。小脳形成不全 (IaとIc)、髄鞘形成の遅延 (Ie、IIa、IIb) 異常小頭や大脳の萎縮 (Ia、Ic、Id、Ie)が見られる。先天性不全症は時々軽いことがある。ほとんどの患者で摂食障害があり、成長が悪い。斜視、異常脂肪分布、乳房退縮が通常的である。
小児のCDG-Ia患者は生後数年で20%が死んでしまうが、小児期を経ると安定化する。実際的にかなり大多数が生存することは、多くの成人CDG患者は未診断のままであることを意味している。

CDGに対する限られた治療の選択肢
CDG-Ibに対しての有効な治療はマンノースの経口投与である3。マンノースはフルクトース-6-P→マンノース-6-Pブロックを迂回し、枯渇したGDP-マンノースプールを補給する。マンノースは低血糖とアンチスロンビンⅢ不足を数週間以内に上昇の方向に、1-2ヶ月以内に血漿性タンパク質レベルを正常化させ、タンパク質-喪失腸症を消失させる。CDG-Ibと証明された成人患者は、誰も現在マンノースを飲んでいないことから、一生飲み続ける必要はないらしい。あるCDG-IIc患者はフコースサプルメントを処方され、シアリルルイスXの生合成に必要なものが供給されたことで、彼の増加していた末梢血好中球数は減少した4。
筋肉細胞表面のキーとなる分子はジストロフィン複合体の一部分であるα−ジストログリカンであり、それは細胞外マトリックスと細胞内骨格の橋渡しをしている。α−ジストログリカンは特殊なタイプの、マンノースが基本となった糖鎖を含んでいる。このマンノースの研究は長いこと無視されてきた(日本以外では!)。これらの糖鎖はN-結合糖鎖合成経路の一部ではないが、糖鎖がα−ジストログリカンの短い領域中でクラスター化し、重要な安定化のためのマトリックスとの相互作用の多くを仲介している。今となっては何種類かの筋ジストロフィーが、これらの糖鎖合成に必要な遺伝子の変異の結果であったことは、驚くに当たらない。筋肉−眼−脳症、福山型先天性ジストロフィー (FCMD)、Walker-Warburg症ではこれらの遺伝子の変異がある。多くの組織、細胞中の全ての型のα−ジストログリカンの糖鎖形成を研究することはこれらの状態が互いにどう異なるのか理解する上で重要であろう。さらに驚くべきことは、成人になって発症する筋ジストロフィーの一つである遺伝性封入体筋炎-II型は、α−ジストログリカン糖鎖中の糖の一つに対して普遍的活性型供与体であるCMP−シアル酸の、生合成に関わる酵素の変異によるものであることが分かった。
糖鎖と糖鎖の組立て工程は非常に複雑である。従って実際的な事として今後もっと多くの病気が発見されることは確かなことである。最も起こり得る領域はO-GalNAc結合糖鎖、グリコサミノグリカン、そしてゴルジで組立て、組織化されるタンパク質である。これらの合成経路の多くには予備経路があり、時には組織特異的に分布する酵素が重複しているので、同定することはより難しいと思われる。良い例としてはヘパラン硫酸合成における多発性遺伝性外骨腫 (MHE)と、ルイスXとルイスY糖鎖を合成する種々フコシルトランスフェラーゼの選択的な発現である。これに比べ、N-結合型経路の生合成前駆体段階での欠損を見つけることは、各ステップとも一つの酵素によって直線状に配列されてくるので容易である。さらにトランスフェリンがN-型糖鎖形成の欠損の強力な分析法を提供してくれる。他の経路については同様な優れた診断法がない。
糖鎖生物学は2001年にはシンデレラサイエンスと呼ばれた。遺伝性糖鎖疾患学は急激に進歩し、特に、もし糖鎖形成を基礎とする治療法が次々と見つかれば、カボチャの馬車は救急車になるであろう。細胞表面のシグナル複合体を構築する糖鎖の役割を理解し、細胞中の糖鎖供与体をいかに増強するかを見つけることが、これらの疾患を理解して治療する助けとなると思われる。将来多くの、より欠損があるものが見つかって来るであろう。
謝辞
本研究はNIH、March of Dimes財団、CDG家族ネットワーク財団からの助成で行われた。



