
Eva Turley
Eva Turley is a Cell Biologist who discovered and cloned RHAMM and is an expert in the area of hyaluronan, hyaluronan receptors and signaling pathways regulating cell motility and invasion. She also has extensive experience in the development of RHAMM and hyaluronan therapeutic reagents for pharmaceutical companies and holds over 15 patents in this area. She is currently a Senior Scientist at The Hospital for Sick Children and Professor in the Department of Laboratory Medicine and Pathology and Department of Anatomy and Cell Biology at the University of Toronto.

Rene Harrison
Rene Harrison has done research in several fields, including cell and developmental biology, with emphasis on signaling and cytoskeletal dynamics. She completed her BSc at the University of Winnipeg and her MSc at the University of Manitoba. She is currently completing her doctoral studies with Dr. E. A. Turley and holds a Medical Research Council of Canada Studentship. She plans to continue working on intracellular signaling dynamics of hyaluronan receptors.
RHAMM belongs to a heterogeneous group of proteins designated hyaladherins, that are linked by their common ability to bind hyaluronan. These proteins can be grouped both according to their extracellular and subcellular distribution and according to the sequences by which they bind hyaluronan, (Fig.1).1
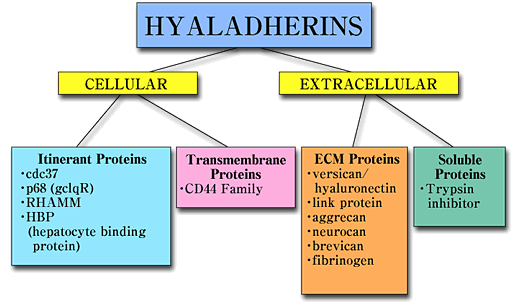
Fig. 1 Hyaladherin family of proteins.
Hyaladherins are a disparate group of proteins linked by a common ability to bind to hyaluronan. They can be grouped according to their cellular and extracellular location. RHAMM belongs to the group of hyaladherins that appear to occur at several locations and which have therefore been designated itinerant hyaladherins.
Hyaladherins occur in the extracellular matrix, on the cell surface and intracellularly. Most of the known hyaladherins couple to hyaluronan via a 100 amino acid "link module". The hyaladherins such as RHAMM are a subgroup that bind to hyaluronan by a 9-11 amino acid sequence containing multiple basic amino acids.1 Most of the characterized hyaladherins in this latter group are intracellular proteins, (Fig.2),1,2 but RHAMM is unique since it occurs as multiple forms, intracellularly and at the cell surface.2-4 However, RHAMM has proven to be one of the more elusive hyaladherins in terms of understanding the molecular rationale for its subcellular compartmentalization and the conditions and mechanisms that generate its multiple protein forms. This protein, originally isolated from supernatant medium of non-confluent embryonic chick heart fibroblasts, is either shed or secreted by the sparsely cultured cells.1 The nascent protein does not contain a signal peptide or a transmembrane domain. Therefore, it may be exported from the cell via chaperone protein(s) and incorporated onto the cell surface via associations with integral and GPI-linked proteins.
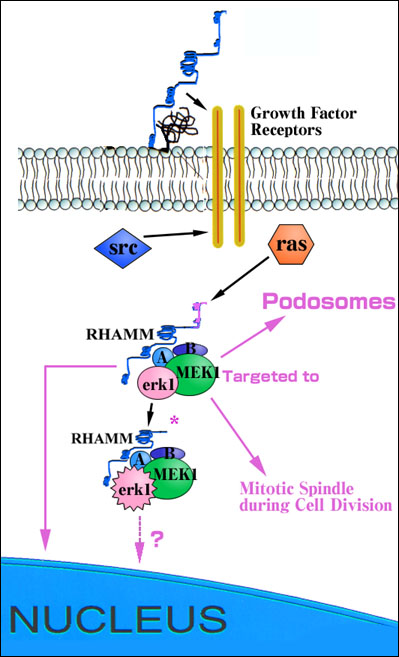
Fig. 2 Diagram of proposed role of RHAMM in regulating the ras/erk1 and src signaling pathways.
Cell surface RHAMM is proposed to modify erk1 activation via an association with growth factor receptors such as the PDGF receptor. Intracellular RHAMM forms are erk1 binding proteins and act downstream of both src and ras at the level of MEK1 to control signaling through ras. RHAMMv4 enhances the activation of erk1 possibly by bringing MEK1/erk1 and unidentified accessory protein (A, B) complexes closer to upstream activators and downstream substrates in dynamic structures such as podosomes and mitotic spindles.
Antibody or peptide blocking experiments, as well as use of dominant negative mutations of intracellular RHAMM forms, indicate that both the cell surface and intracellular forms of RHAMM are required for cell motility and cell cycle progression through G2M. Intracellular RHAMM forms are erk 1 (from extracellular regulated kinase) binding proteins while cell surface RHAMM forms appear to act as co-receptors that modify signaling through integral proteins such as the PDGF receptor. One intracellular form of RHAMM (v4) is transforming when overexpressed.1,5,6
The constitutively expressed and most common RHAMM mRNA transcript encodes the largest intracellular RHAMM protein, 85 kDa (human) and 95 kDa (murine), that has been designated v5, (Fig.3 and 4). This RHAMM protein reacts with antibodies 1,2 and 3 (Fig.5). Evidence is accumulating for the existence of multiple alternative spliced variants of the v5 form, (Fig.3 and 5).4,7,8 Further, shorter RHAMM forms (e.g. v4, Fig.3), which react only with Ab 2 and Ab 3 (Fig.5), and represent N-terminal truncations of v5, appear soon after cells are plated, or following tissue injury, or upon activation of the small GTPase, ras (from rasheed sarcoma virus). These proteins of 60-73 kDa have been reported by many laboratories to represent minor forms of RHAMM protein.
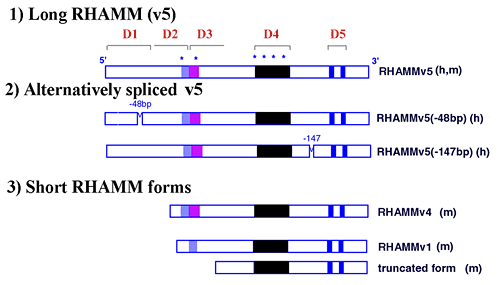
Fig. 3 Predicted domain structure of RHAMM in human (h) and mouse (m).
Functional domains (D1-D5) have been identified by structure/ function analysis and are required for RHAMM-mediated cell motility and passage through the cell cycle. D1 is a novel protein domain that negatively regulates the ability of RHAMM sequence to promote activation of erk1 kinase; D2 encodes an imperfect leucine zipper that is required for RHAMM-mediated cell motility and podosome formation; D3 is a novel sequence that is required for interaction of intracellular RHAMM with MEK1; D4 is a novel sequence that is repeated up to 8 times in the murine protein and contributes to the binding of erk1 to intracellular RHAMM; D5 encodes hyaluronan binding motifs that are responsible for interaction of hyaluronan with cell surface RHAMM and erk1 binding to intracellular RHAMM. RHAMM, like CD44, has been reported to exist as multiple forms, some of which are generated by alternative splicing of the longest RHAMM (v5) mRNA transcript. Several shorter, activated forms of RHAMM have also been reported, and these may be generated either by separate mRNA transcripts, internal start codon usage of the v5 transcript, or proteolysis of the v5 protein.
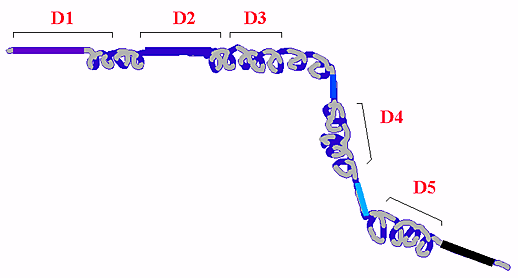
Fig. 4 Predicted secondary and domain structure of RHAMM.
Secondary structure analysis indicates RHAMM exists largely as a coiled coil protein punctuated by small non-coiled coil stretches often preceding functional domains.
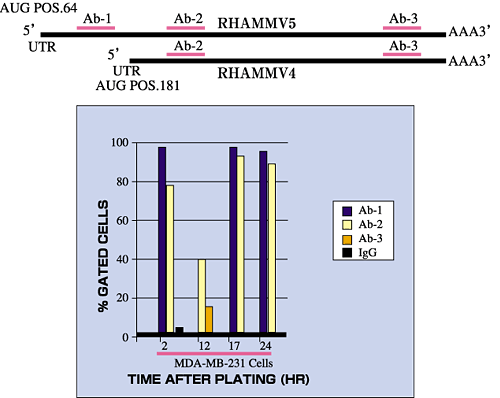
Fig. 5 RHAMM forms are regulated in their expression.
Cell surface RHAMM expression (detected by 3 different RHAMM antibodies) in a malignant breast cancer cell line (MDA-MB 231) fluctuates with time after cell plating. Transformed fibroblasts express two major RHAMM forms that correspond to v5 (95 kDa) and v4 (73 kDa), as identified by molecular weight and antibody reactivity. The 73 kDa form does not encode the N-terminal sequence found in v5 and is therefore thought to represent v4.
V5 is translated from a full length, commonly expressed mRNA transcript that is detectable in most tissues. A full length RHAMM mRNA transcript encoding v4, (Fig.3), has also been described2 that has a 5' untranslated region distinct from the v5 transcript. The v4 transcript is minor and expressed early after cell plating. However, it is possible that the v4 protein is also generated by additional mechanisms such as usage of internal start codons present within the v5 mRNA transcript, or proteolysis of the v5 protein.
RHAMM is an unusual protein in that much of its sequence does not resemble that of functional domains common to either intracellular or cell surface proteins known to be involved in regulating signaling of cell motility or cell cycle. Therefore, extensive structure-function analysis of RHAMM sequence has been done to identify the key regions required for these biological effects. A challenge for the future will be to identify RHAMM-binding proteins to provide a clearer picture of the mechanisms by which this hyaladherin controls cell behavior.
RHAMM sequence from amino acids 163 to the carboxy-terminus is largely a coiled coil with non-coiled coil regions punctuating this secondary structure, (Fig.5). The coiled coil structure may permit self-association, which would provide a rationale for the remarkable effectiveness of mutant RHAMM forms to act as dominant negative function suppressers.2 D2-D5 domains, (Fig.4), are each required for RHAMM-promoted cell motility and for passage through the cell cycle. Deletion or mutation of any one of these domains is sufficient to ablate the ability of RHAMM to signal and the ability of RHAMM overexpression to transform fibroblasts.1 D1 negatively regulates these functions of D2-D5 and therefore v5 represents an inactive form of RHAMM, while the truncated forms, such as v4, represent the active form. RHAMM thus resembles oncogenes such as raf, which are also activated by removal of peptide sequences.
The N-terminal D1 domain unique to v5 that negatively regulates the function of downstream RHAMM sequences is an entirely novel sequence, and the manner in which it regulates the function of D2-D5 is not yet clear. It is characterized by the presence of a src (from sarcoma) homology 3 binding domain (SH3) and multiple erk 1 phosphorylation sites. It is possible that D1 may cover at least one of RHAMM's downstream domains by binding to an accessory, regulatory protein. Alternatively, the SH3 binding domain may place v5 in a separate sub-cellular compartment from v4, restricting access of erk1, for instance, to key substrates that are required for signaling motility and cell cycle progression.
D2 encodes an imperfect leucine zipper, and has been shown to permit binding of cell surface RHAMM to fibronectin in the extracellular matrix, an interaction that is required for the formation of podosomes, enhancement of cell motility, and release of metalloproteinases.6
D3 of intracellular RHAMM mediates an association between RHAMM and MEK1, forming a RHAMM/MEK1/erk1 complex detected following immunoprecipitation of RHAMM.3 This interaction is indirect since MEK does not bind to RHAMM in vitro. Interestingly, D3 encodes a 6 amino acid sequence, VSLEKEL, that is present in another MEK1 binding protein, MP-1.
D4 is a 21 amino acid sequence repeated up to 8 times in murine RHAMM, and is required for full binding of erk1 to RHAMM. Antibodies to this domain added to the culture medium promote cell motility and focal contact turnover, mimicking the effect of hyaluronan1 and indicating that this domain is also important to the function of cell surface RHAMM. This domain has been classified with a group of 91 proteins that are involved in signaling.
The first reported functional domains of RHAMM were its hyaluronan binding motifs present in D5, which encode sequences of basic amino acids that are also common to other intracellular hyaladherins, such as cdc37 and p68.1 These domains are the sole hyaluronan-binding sites of RHAMM and are key to generating a motility signal from exogenous hyaluronan.2,9 On intracellular RHAMM forms, D5 mediates binding of erk1 to RHAMM. It therefore appears that cell surface RHAMM binds to hyaluronan via the D5 domain, while intracellular forms of RHAMM utilize this site to bind to erk1. This is consistent with evidence that mutations in this domain in intracellular RHAMM forms block activation of erk1.2
Sugar transport signatures are present in the N-terminus of v5 and in the carboxy-terminal sequence common to all RHAMM forms, and may be required for transport of hyaluronan into the cell.10
Furthermore, a cyclin signature present in all forms is consistent with the involvement of RHAMM in the cell cycle.5 However, the protein partners and the precise molecular function of these intriguing homologies are not yet clear.
Finally, RHAMM contains many potential sites for post-translational modifications, including N-glycosylation sites, myristoylation sites, and notably, multiple serine-threonine phosphorylation sites. The effects that these modifications might have on subcellular localization and protein interactions remain to be determined.
Subcellular Distribution
RHAMM has been reported by several laboratories to occur at the cell surface, within the cytoplasm and in the nucleus.1,11 Interestingly, the biologically active v4 form, localizes primarily to dynamic subcellular structures such as podosomes and rapidly turning over (e.g., non-acetylated) microtubules, and to dynamic microtubular structures such as mitotic spindles, (Fig.6). In contrast, v5 occurs primarily in the perinuclear region. Scanning electron microscopy indicates that cell surface RHAMM, whose molecular nature is not yet known, is located primarily in extending microvilli.6 The expression of the cell surface form of RHAMM is particularly ephemeral, fluctuating in expression following cell plating and gradually disappearing as cell-cell contact increases.3,6 Addition of molecules such as hyaluronan, however, promotes the rapid appearance of RHAMM at the cell surface, likely from caveoli, (Fig.7).3 These last data suggest that some intracellular RHAMM must be stored, ready for targeting to the cell surface.
Cell surface RHAMM appears to be less dynamic and more constantly expressed with disease. For example, cell surface RHAMM can always be detected on the surface of aggressive subsets of multiple myeloma cells, breast cancer cells,4 and at a low level on macrophages isolated from the synovial fluid of patients with rheumatoid arthritis. Many of the alternatively spliced forms of RHAMM shown in Fig.3 have been detected in transformed cells or cancer biopsies, although their subcellular distributions remain uncharacterized.
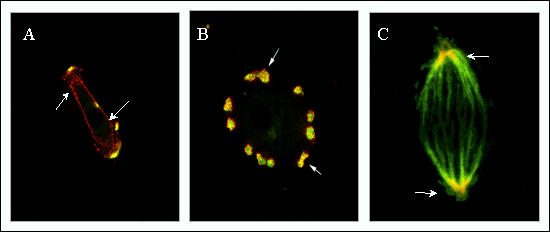
Fig. 6 RHAMM occurs in transient structures, and its expression is upregulated at the cell surface during disease processes.
(A, B) Dual immunostaining of RHAMM and cortactin, a marker for podosomes, which are localized sites of protease release. Early after cell plating (A, 2 hour) RHAMM is primarily localized at the cell membrane but is recruited into developing podosomes by 8 hours (B) after cell plating. (C) RHAMM and tubulin show co-localization within mitotic spindles.
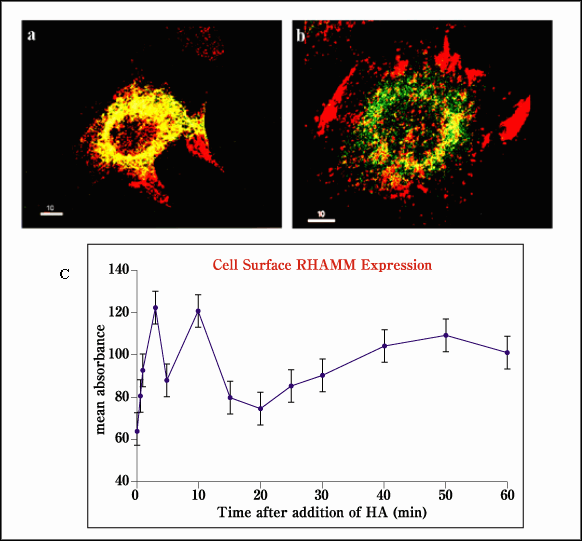
Fig. 7 Cell surface RHAMM is "flashed" onto the cell surface in response to exogenous hyaluronan.
(a) The colocalization of RHAMM (red) and caveolin (green) suggests that some intracellular RHAMM is stored in caveoli. (b) The addition of hyaluronan to cells causes a rapid redistribution of RHAMM from caveoli onto the cell surface as detected by FACs (c, Ab-3).
Cell Type Distribution
RHAMM forms are expressed in most normal avian and mammalian cell types. These include fibroblasts, smooth muscle cells, endothelial cells, macrophages, immature thymocytes, B cell lineages, bone marrow stromal cells, keratinocytes, sperm, astrocytes, astrocytomas, central nerve cells, microglial cells, sprouting olfactory nerve cells, lung adenocarcinoma, malignant pancreatric cells, and malignant breast epithelial cells. In general, the highest expression of cell surface RHAMM and small RHAMM forms (e.g., v4) is associated in the above cell types with early plating after subculture, when cells are forming extensions,12,13 initiating cell motility, and entering into cell cycle.
Tissue Distribution
A complete study of RHAMM protein expression in different adult or embryonic tissues has not been published, but unreported accounts utilizing RT-PCR analyses indicate that v5 is present in most tissues. A few reports of immunocytochemical analyses from a limited number of tissues suggest that RHAMM expression may vary with specific cell types within tissues. For instance, RHAMM is expressed in the highly responsive, branching locus coeruleus of the rat brain, relative to other brain cell types,12,13 and in migratory medullary thymocytes of the developing thymus.14
A peculiar feature of RHAMM-regulated signaling is that it activates specific signaling pathways, yet controls the extent to which these same pathways can be further activated by growth factors such as PDGF.3,6 A key target of RHAMM-regulated signaling appears to be the cytoskeleton. For example, the ability of RHAMMv4 to disassemble focal contacts, which are sites of integrin and growth factor clustering, may help to restrict integrin and growth factor signaling.
RHAMMv4 promotes signaling through at least two molecular switches, src and the small GTPase p21 ras, (Fig.2). Both of these molecules can function as oncogenes and have been shown by others to regulate cytoskeletal assembly, proliferation and motility. In particular, src contributes to a motility signal in response to growth factors, a property that requires its tyrosine kinase activity, and that also mediates changes in the actin cytoskeleton. Cell surface RHAMM interacts with both hyaluronan and fibronectin to modify tyrosine phosphorylation of key actin and integrin binding proteins such as focal adhesion kinase (FAK) and cortactin, events which are associated with focal adhesion turnover.2 Changes in protein tryosine phosphorylation are mediated by src, which is transiently activated by cell surface RHAMM and which associates with RHAMM in cell lamellae where focal adhesions are primarily located.9 Cell surface RHAMM-controlled src activity may also contribute to the ability of this hyaladherin to modify signaling through growth factors.3,6
In addition to its effect on src, both cell surface and intracellular RHAMM forms are required for signaling through p21 ras, (Fig.2). Ras controls multiple downstream pathways including at least three map kinase pathways. Interestingly, RHAMM appears to act on only one of these, the erk1 kinase pathway, and this interaction is required for the ability of v4 to signal motility and proliferation. All intracellular forms of RHAMM can bind directly to erk1 kinase and most forms interact indirectly with the upstream kinase activator, MEK1.3 However, only the short forms of RHAMM, notably v4, strongly activate erk kinase 1, acting to enhance the function of MEK1. V4 co-distributes with erk1 in mitotic spindles and in podosomes, (Fig.8), and these associations appear to be required for the ability of microtubules to turnover on the one hand, and for the stability of podosomes on the other.
A function of v4 may be to localize erk1/MEK complexes to upstream activators and accessory proteins in specific, dynamic cytoplasmic structures, (Fig.2). V4 overexpression constitutively activates erk1 kinase 4-5 fold above background, and this level of activation is sufficient to enhance AP-1 formation6 and expression of AP-1 regulated genes such as MMP-9. Although RHAMM v4 activates erk1 4-5 fold above quiescent levels, it restricts further activity of erk1 by pro-inflammatory cytokines such as IL-1, TNF-alpha and PDGF, which can promote erk activation by up to 20 fold in the absence of RHAMM.6
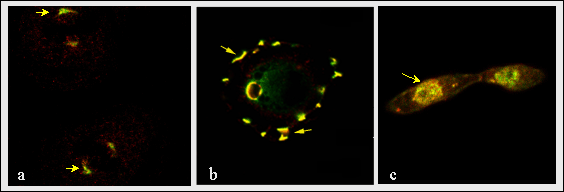
Fig. 8
Colocalization of hemaglutinin tagged RHAMMv4 and erk in mitotic spindles (a), podosomes (b) and nucleus (c) (arrows). The colocalization of both molecules predicts a key role for v4 in localizing this map kinase to dynamic cytoplasmic structures. Erk, for instance, phosphorylates a variety of substrates in mitotic spindles, and RHAMM may play a key accessory function permitting localized access of erk to its substrates in the spindle.
As noted above, multiple RHAMM forms have been reported to be overexpressed in disease while the v5 form appears to be constitutively expressed in most adult homeostatic tissues. It is fascinating, therefore, that the shorter forms of RHAMM have recently been shown to appear transiently in normal tissue that is responding to injury, for example following bleomycin-induced damage of lung or following excisional injury of skin. In rat skin excisional wounds, RHAMMv4 is expressed rapidly after the injury but disappears by 24 hours, (Fig.9). During that time, RHAMM is elevated in keratinocytes, macrophages and migrating fibroblasts.
Consistent with these observations, transgenic expression in keratinocytes, of a function suppressing mutant form of v4 that is unable to bind to erk1 or to complex with MEK1, reduces activation of erk1 and alters the course of excisional wound repair, notably restricting contraction and re-epithelialization of the wound, (Fig.10). These effects of RHAMM on wound repair appear to be pleiotropic since application of RHAMM peptides to injured rodent skin results in reduction of contraction, consistent with the transgenic model, but also inhibits collagen III synthesis resulting in a reduction in scar formation, (Fig.10). Some of these effects appear to be due to reduced fibroblast and macrophage migration into the wound site. Application of RHAMM peptides at the time of bleomycin insertion into rat lungs also prevents macrophage influx, resulting in a marked reduction in fibrosis of the lung tissue. The strong upregulation of RHAMM following wounding, chronic inflammation and cell transformation predicts an involvement of growth factors or oncogenes in control of the expression of this hyaladherin, consistent with reports that TGF-beta and mutant active ras enhance expression of RHAMM protein forms.
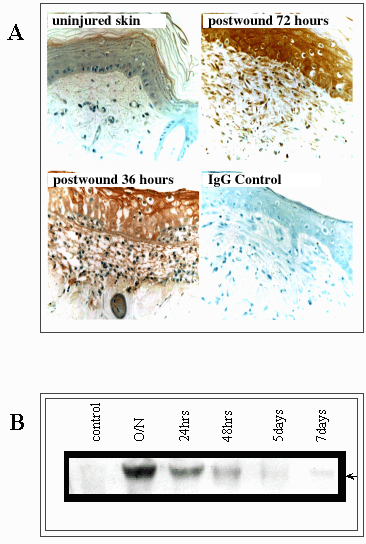
Fig. 9 RHAMM expression, particularly that of the activated 73 kDa (v4) is transiently elevated following tissue injury.
(A) Immunostaining for RHAMM in an excisional skin wound shows an upregulation of this itinerant hyaladherin in keratinocytes, infiltrating macrophages and invading fibroblasts. (B) This cellular influx in response to injury coincides with a transient appearance of the 73 kDa RHAMM protein.
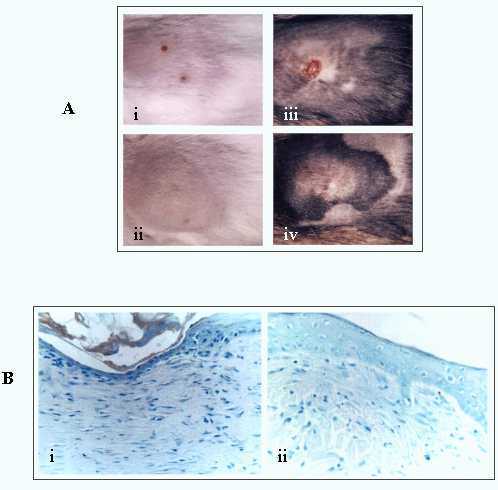
Fig. 10
(A) The course of wound repair is modified in transgenic mice expressing a dominant negative RHAMMv4.
(i, ii) represent wounds of wild-type mice at 3 days (i) and 1 week (ii) after excisional injury. (iii, iv) represent the transgenic mouse at the same time after injury. Ablation of RHAMM function blocks wound contraction. (B) Similarly, a RHAMM peptide mimetic applied to an excisional wound blocks the contraction (ii) observed in a control (i).
The above data are consistent with a model where transient, activated forms of RHAMM (60-73 kDa) contribute to regulated stages of cell activation in tissue following injury, (Fig.11). Cells in tissues are generally maintained in a homeostatic state of differentiation that is interrupted at regular intervals by cell turnover. After insults such as injury or somatic mutation, cells are activated to respond, and it is reasonable to propose that this activation is normally tightly controlled. For instance, following tissue injury, the influx of plasma and local adherence of platelets results in a cytokine storm that, if cells were immediately and maximally responsive to these factors, would result in a chaotic and uncontrollable repair process. Expression of activated RHAMM and cell surface RHAMM forms would result in a transition injury state characterized by the appearance of podosomes, a regulated activation of erk1 kinase to permit expression of some metalloproteinases, and a restricted ability to respond to growth factors. This state would permit initiation of matrix remodelling, cell motility and initiation of cell cycling. Partly as a result of extracellular matrix remodeling, some white blood cells would be attracted to the injured area, providing a future source of additional cytokines/growth factors that would ultimately facilitate the progression and escalation of the repair process, following disappearance of RHAMM and other transitional molecules. Thus, RHAMM expression in the transition stage of injury would control the rate at which cells progress to a highly cytokine-responsive state, and its disappearance would permit increasing responsiveness, as clustering of integrins and the formation of focal adhesions proceed.
This model predicts that the transiently expressed RHAMM forms, particularly those expressed at the cell surface, would provide excellent therapeutic targets to limit the extent of fibrosis and scarring of repairing tissue without toxicity to uninjured tissues, which do not express these RHAMM forms. Certainly, data showing a reduction of scarring following excisional injury of skin in the presence of RHAMM peptides provide support for this possibility.
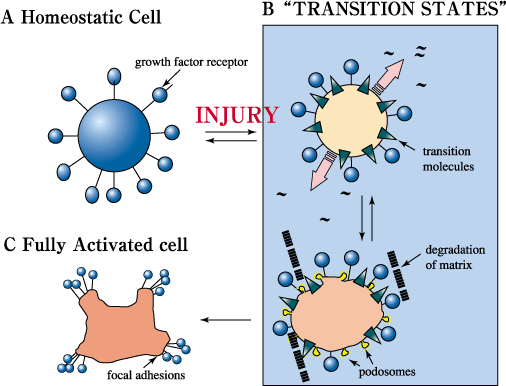
Fig. 11 A model of the proposed role of RHAMM and itinerant hyaladherins during response to injury.
Following injury, when cells are exposed to cytokine/growth factor "storms", cells are predicted to be activated in a tightly regulated process that involves several intermediate steps. This permits a regulated progression towards a fully activated cell that is highly responsive to cytokines/growth factors. In an injured environment, RHAMM and other transitional molecules are thought to play the dual role of participating in the initial activation of cells, (e.g. podosome production, initiating matrix remodeling, growth factor activation, and attraction of white cells) and controlling the extent to which injured cells can respond to cytokines/growth factors. RHAMM thus contributes to a rigid control of the injury response. As active RHAMM forms disappear, cells are able to proceed to a more highly responsive state to achieve efficient repair. Once this has been accomplished, the maximally activated state is repressed. Consequently, maintenance of high levels of active RHAMM forms is proposed to contribute to disease processes such as neoplasia.



