
Yasuhiko Kizuka
Professor at the Institute for Glyco-core Research (iGCORE), Gifu University
2004 Faculty of Pharmaceutical Sciences, Kyoto University. 2009 Ph.D., Pharmaceutical Sciences, Kyoto University. From 2009 to 2017 (8.5 years), he was a postdoctoral researcher in the Taniguchi group at RIKEN. From 2017 to 2022, he was an associate professor at the G-CHAIN center of Gifu University, and from 2022–, Professor at iGCORE, Gifu University. His research focuses on understanding the regulation of expression and activity of glycosyltransferases using biochemical approaches.
Elucidating the pathological mechanisms of Alzheimer’s disease (AD), the most common form of dementia, and developing new methods for its early detection are crucial. In this review, the known relationships between AD and glycans are presented with a particular focus on comprehensive glycan analysis and the roles of specific glycans in AD. Summarizing these studies, the potential development of glycan-targeted diagnostic and therapeutic approaches for AD are discussed.
Japan has become a super-aging society, with approximately 29% of the population currently over 65 years old, a figure projected to rise to 40% by 2070 (Cabinet Office, Government of Japan, 2024 Annual Report on the Aging Society, https://www8.cao.go.jp/kourei/whitepaper/w-2024/html/zenbun/index.html). In this aging society, dementia poses a significant threat to individuals and represents a major burden on national healthcare and the economy. As the most prevalent form of dementia, Alzheimer's disease (AD) remains a critical medical challenge to solve, highlighting the urgent need for effective diagnostic and therapeutic strategies.
AD is a progressive neurodegenerative disease characterized by impairments in memory and cognition1,2. Although antibody drugs that ameliorate disease progression are under development, no fundamental cure has been reported. One of the major challenges in AD therapy is the long preclinical phase, with pathological changes occurring over approximately 20 years before clinical symptoms appear. These changes, as described below, are already in progress when clinical symptoms manifest, and thus, early diagnosis at the pre-symptomatic stage is crucial for effective intervention3,4. In addition, although some genetic risk factors have been identified, most AD cases are sporadic, with various factors making the pathogenesis highly complex1,2. Therefore, the molecular mechanisms underlying AD remain mostly unresolved. In this review, potential diagnostic and therapeutic approaches for AD will be presented, with a focus on the role of glycans in AD. Recent advances in comprehensive structural analysis of glycans and glycopeptides have improved our ability to study glycan-related changes in AD. In the following section, the current state of comprehensive glycan analysis in AD research will be presented. That section will be followed by a discussion on specific AD-related glycans: bisecting GlcNAc (one of our research targets), O-GlcNAc, GM1 ganglioside and O-GalNAc-type glycans.
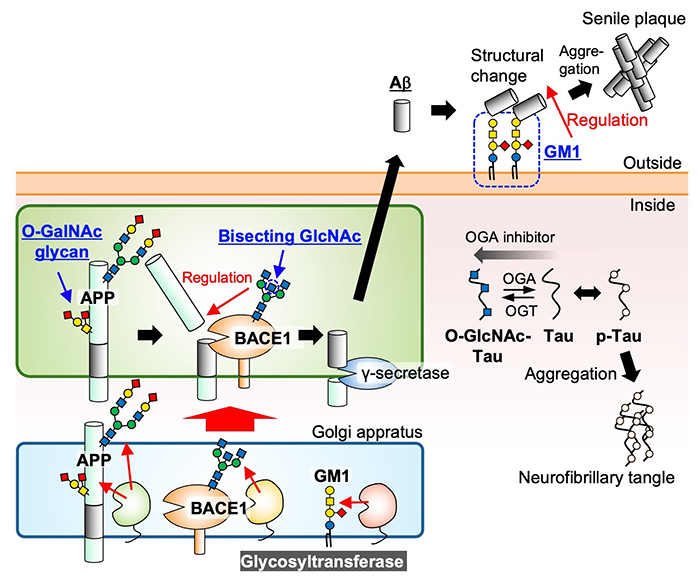
Two major pathological hallmarks are observed in the brains of AD patients: “senile plaques” and "neurofibrillary tangles"1,2. Senile plaques are aggregates of extracellular peptide amyloid-β (Aβ), and neurofibrillary tangles are intracellular aggregates of the abnormally phosphorylated tau protein (Figure 1). Although a clear causal relationship between Aβ and tau accumulation has not yet been clarified, the cascade hypothesis that Aβ deposition is upstream and causes AD is widely accepted5 because Aβ deposition precedes tau accumulation1,2 and all causative genes for familial AD encode proteins involved in Aβ production and accumulation6. Aβ is produced from the precursor protein APP (amyloid precursor protein) by stepwise cleavage involving BACE1 (beta-site APP cleaving enzyme-1) and γ-secretase (Figure 1)7. In addition, Aβ and tau are localized in different intracellular compartments. Aβ is produced in the lumen of secretory organelles, such as endosomes, and secreted, whereas tau is essentially localized to the cytosol (recently, secreted tau has also attracted attention). Thus, proteins involved in Aβ production and aggregation face the luminal side of secretory organelles or outside the cells, whereas tau-related proteins face the cytosolic side. In terms of glycosylation, Aβ-related proteins are likely modified with N- and O-glycans, whereas tau-related proteins (including tau) likely undergo O-GlcNAc modification. In this review, several glycans attached to glycoproteins will be highlighted, and the relationship between a ganglioside GM1 and AD will be discussed.
Recent advances in mass spectrometry (MS)-based techniques for comprehensive glycan structural analysis have enabled detailed analysis of overall glycans in cells, tissues and proteins. These methods can be applied to large-scale cohort studies, providing valuable insights into glycan-related biological processes. MS-based analytical methods can be broadly classified into glycomics and glycoproteomics. Glycomics involves the cleavage of glycans from a sample, allowing for the structural analysis of glycans. In contrast, glycoproteomics enables the identification of both glycan structures and their specific attachment sites within the amino acid sequence of glycoproteins by digesting proteins into glycopeptides while preserving intact glycans. Although glycoproteomics provides more comprehensive information, a current technical limitation is that the analyzable glycans are often restricted to N-glycans in many cases.
Recently, glycomics and glycoproteomics have been conducted to analyze glycan alterations in AD, and these approaches are expected to be applied in the future to developing glycan-targeted AD biomarkers. Cerebrospinal fluid (CSF) is frequently used as samples for analysis, whereas blood and postmortem brain samples are also used in some studies. In more detail, recent glycomic analysis of CSF from dozens of healthy individuals, mild cognitive impairment (MCI) and AD patients8,9 identified alterations in several glycans. Notably, an increase in glycans with bisecting GlcNAc (as described below) and a decrease in sialic acid were commonly observed in MCI and AD patients compared with healthy individuals8,9. Moreover, an increase in fucosylation was reported in another study10. A liquid-chromatography (LC)-MS glycomic analysis of postmortem AD brains detected changes in various glycans; however, no common glycan structural motifs were identified among the altered glycans11.
In glycoproteomic analysis, Chen et al. identified nearly 3,000 glycopeptides in CSF from healthy individuals and AD patients, reporting changes in N-glycan structures for specific glycoproteins derived from AD samples12. Similar analyses using brain samples from AD patients have further expanded this understanding. Suttapitugsakul et al. identified over 2,000 glycopeptides and reported overall and site-specific glycan profile changes in AD13. In 2024, more than 12,000 glycopeptides were identified in the brains of AD patients, revealing more detailed site-specific glycan changes14. In addition to glycan structures, another LC-MS analysis using AD brain samples revealed differences in the presence or absence of N-glycans on proteins and variations in the glycosylation efficiency at the same site15. As comprehensive glycomics and glycoproteomics advance, the information identified will continue to grow rapidly. Future large-scale cohort studies with extensive sample sizes are expected to provide even greater detail and deeper insights into the role of glycans in AD.
Bisecting GlcNAc is the central GlcNAc residue attached to β-Man of N-glycans and is produced by an enzyme designated GnT-III (MGAT3)16. Bisecting GlcNAc was reported to inhibit the formation of other branches and terminal structures of N-glycans17,18 and to suppress cancer proliferation and metastasis19. Among animal tissues, expression of GnT-III is highest in the brain, especially in neurons20. Although the roles of bisecting GlcNAc in the brain remain unknown, in 2010, Dr. Endo’s group analyzed human brains and demonstrated that the mRNA level of MGAT3 in AD patients is higher21. This observation suggested a relationship between bisecting GlcNAc and AD, and thus, we analyzed how AD pathology is altered in GnT-III-deficient mice.
Mouse AD models are typically used in AD research because mice do not spontaneously develop AD22. Thus, we crossed human APP transgenic mice with a familial AD mutation (hereafter APP-Tg), a frequently used model in AD research, with GnT-III-deficient mice. We observed that the number of senile plaques in the brain was drastically reduced in GnT-III-deficient APP-Tg23. Furthermore, in a Y-maze behavioral test for cognitive performance, the poor performance of APP-Tg mice was restored in GnT-III-deficient APP-Tg mice and to performance levels similar to that of wild-type mice. These findings indicate that loss of bisecting GlcNAc reduces Aβ accumulation in the brain and improves AD pathology.
The mechanism by which loss of bisecting GlcNAc reduces Aβ levels remains an important question. As described above, Aβ is produced through a two-step cleavage of APP (Figure 1), and most proteins in the APP metabolic pathway undergo N-glycosylation24. Glycan analysis of these glycoproteins revealed that BACE1, the enzyme responsible for the first cleavage of APP, is heavily modified with bisecting GlcNAc. In GnT-III-deficient APP-Tg mice, the amounts of cleavage products generated by BACE1 were reduced significantly, indicating that the loss of bisecting GlcNAc impairs BACE1 function. Further analysis of BACE1 features revealed that while the enzymatic cleavage activity of BACE1 was not affected, its subcellular localization was altered. Specifically, the BACE1 location shifted from early endosomes to late endosomes and lysosomes because of the loss of bisecting GlcNAc. Because early endosomal localization of BACE1 was reported to be pivotal to APP cleavage by BACE125,26, this change in BACE1 localization may be the primary cause of reduced Aβ production. In conclusion, bisecting GlcNAc positively regulates Aβ production by controlling the localization of BACE1 and may serve as a new therapeutic target for AD. However, the underlying mechanism by which the loss of bisecting GlcNAc alters BACE1 localization remains unclear and requires further investigation.
As described above, the level of bisecting GlcNAc was found to increase in MCI and AD patient CSF samples, as determined by glycomic analysis8,10. Furthermore, the detection of bisecting GlcNAc in CSF using a lectin-based ELISA system shows that this glycan can be a biomarker for diagnosing AD and early cognitive decline27,28. Another study showed that bisecting GlcNAc in blood may also be used to diagnose AD in combination with tau levels and APOE genotypes29.
O-GlcNAc is a post-translational modification of Ser/Thr residues with a GlcNAc monosaccharide, which takes place in the cytosol, nucleus and mitochondria30. Over 1,000 proteins are O-GlcNAcylated, and in the context of AD, the O-GlcNAc modification of tau has been well studied31,32. Tau is phosphorylated at many sites, and aggregates of abnormally phosphorylated tau are the entity of neurofibrillary tangles33. In general, as both O-GlcNAcylation and phosphorylation occur at common Ser/Thr residues and often compete with each other30,34, it is conceivable that O-GlcNAcylation also regulates tau phosphorylation. Yuzwa et al. reported that an increase in O-GlcNAc levels using an inhibitor of O-GlcNAc degrading enzyme decreases tau phosphorylation31. Furthermore, in mice expressing mutant tau (P301L), increased levels of O-GlcNAc have been shown to suppress tau phosphorylation and aggregation and improve pathology35,36. These findings suggest that increased O-GlcNAc levels may have a therapeutic effect on AD. In contrast, as described above, O-GlcNAc modifications occur widely in numerous proteins, and systemic changes in O-GlcNAc levels may cause functional alterations to many proteins. Therefore, this point should be considered when targeting O-GlcNAc to treat AD.
Although cytosolic tau generally does not undergo N-glycosylation, studies have reported that tau may be abnormally N-glycosylated in AD patients15,37,38. Additionally, secreted tau in the blood is a promising early diagnostic marker candidate; however, the tau secretion pathway is unclear. If future glycoproteomic analyses provide convincing evidence that tau undergoes AD-dependent N-glycosylation, this abnormal glycosylation may represent a research target for developing new therapeutic targets of AD and enhance our understanding of the mechanisms associated with glycosylation and secretion of tau.
GM1 is an AD-related ganglioside that promotes AD pathogenesis by acting as a seed for Aβ aggregation39,40. Conversely, GM1 has been reported to be protective against neurodegeneration in AD41. Dr. Yanagisawa’s group detected Aβ species bound to GM1 in the brains of early AD patients42. Subsequently, many in vitro and in vivo studies suggested that Aβ bound to GM1 functions as a seed for aggregation. For example, monomeric Aβ binds to various gangliosides, including GM143. The GM1–Aβ interaction depends on the composition and cluster state of the lipid moiety of GM144, and NMR analysis showed a conformation shift of Aβ from α-helix to β-sheet upon binding to GM1 clusters, causing Aβ fibrilization45,46. In addition, GM1-bound Aβ was detected in the brains of APP-Tg mice and human CSF47,48. Moreover, enhanced degradation of gangliosides in AD model mice reduced the levels of Aβ bound to gangliosides, leading to improved performance in behavioral tests49. These findings suggest that gangliosides such as GM1 interact with Aβ and facilitate AD pathogenesis.
Studies using mutant mice targeting GM1-related enzyme genes present complex findings and require careful interpretation. In B4galnt1 knockout mice, which lack GM2 synthase B4GALNT1, GM1 disappeared while GM3 accumulated; however, Aβ accumulation increased50. In another study using cultured cells, overexpression of B4GALNT1 caused an increase in BACE1 levels51, suggesting that GM1 may influence both Aβ aggregation and the Aβ production process. In addition, mice lacking ST3GAL5, which is required for synthesizing various gangliosides, including GM1 and GM3, showed significantly reduced Aβ accumulation and improved cognitive performance52. The reason for these complex results is unclear but may be related to the varying affinities of different gangliosides toward Aβ, and that overexpression or deletion of one biosynthetic enzyme may affect the levels of multiple glycolipids.
O-GalNAc-type glycans are major O-glycans that are attached to multiple AD-related molecules. In particular, the O-GalNAc glycosylation of APP has been well characterized, and numerous O-glycosites have been identified in APP from human CSF and cultured cells53,54. The structure of O-GalNAc-type glycans may affect Aβ formation because human APP is O-glycosylated near the BACE1 cleavage site.
The biosynthesis of O-GalNAc-type glycans is initiated by transferring GalNAc to Ser/Thr residues, a process catalyzed by a group of enzymes designated as GALNTs55. Elevated mRNA levels of several GALNTs have been reported in the brains of AD patients56. Overexpression of one of these upregulated enzymes, GALNT6, in cultured cells reduced Aβ secretion into the culture medium. Although the molecular mechanism remains unclear, in vitro assays have shown that GALNT6 transfers GalNAc to an APP-derived peptide close to the BACE1 cleavage site56.
Notably, the APP gene produces different protein isoforms, neuron type (695) and endothelial cell type (770), which exhibit distinct O-glycosylation patterns. APP770 expressed in endothelial cells produces Aβ selectively from the glycoform with O-GalNAc-type glycans57. Additionally, APP770 undergoes modification of O-GalNAc-type glycans during endocytosis from the cell surface58. Given that Aβ deposition in blood vessels frequently occurs in AD patients, leading to cerebral amyloid angiopathy59, the unique glycosylation process of APP in endothelial cells may contribute to AD pathogenesis. Interestingly, an O-GalNAc-type glycan on a Tyr residue of Aβ has also been identified in CSF54. Further research examining the mechanisms of these unique O-glycosylation processes of APP and their roles in AD are required.
TREM2 is a single transmembrane protein expressed in myeloid cells and was found to be associated with AD in a genome-wide association study (GWAS)60. TREM2 is a receptor for various molecules, including APOE, lipids and nucleic acids61. Increased TREM2 expression has been observed in AD patients and mouse models62,63. Interestingly, the AD-associated R47H mutation in TREM2 reduces its ligand-binding activity64 and causes changes in the N-glycan structures of TREM2 with altered sialylation and fucosylation65. As the mutant TREM2 showed enhanced lysosomal degradation, these findings suggest that N-glycans on TREM2 play a crucial role in regulating TREM2 stability and ligand-binding properties, potentially contributing to the pathogenesis of AD.
In this review, previous studies on various glycans implicated in the onset and progression of AD have been summarized. These studies indicate that altered glycans and their biosynthetic enzymes may serve as potential biomarkers for AD diagnosis and that therapeutics modulating the expression and functions of these glycans may represent effective strategies. Recent findings indicate that a large amount of glucosamine is contained in brain glycogen and serves as a UDP-GlcNAc source for glycan biosynthesis66, revealing a novel mechanism underlying glycan expression in the brain. In the future, developing methods to specifically detect AD-related glycans and new compounds or systems capable of selectively modulating glycan biosynthesis and functions will be essential for advancing diagnostic and therapeutic approaches for AD.
The study on GnT-III-deficient mice described in this review was conducted by the Disease Glycomics team at RIKEN, which was led by Dr. Naoyuki Taniguchi. I would like to thank Dr. Taniguchi and Dr. Kitazume for their supervision. I also thank Dr. Takashi Saito (Nagoya City University) for commenting on this review.



