
Kazuki Sato
Junior Associate Professor, Tokyo University of Science
He received a Ph. D. degree from University of Tokyo in 2013 under the supervision of Dr. Takeshi Wada. After working for five years at Toray Industries Inc. as a researcher, he was appointed as an assistant professor at Prof. Takeshi Wada’s laboratory, Tokyo University of Science. He was promoted to Junior Associate Professor in 2023. His research focuses on the synthesis of phosphorus containing biomolecules and their P-modified analogs including carbohydrate and nucleic acids derivatives.
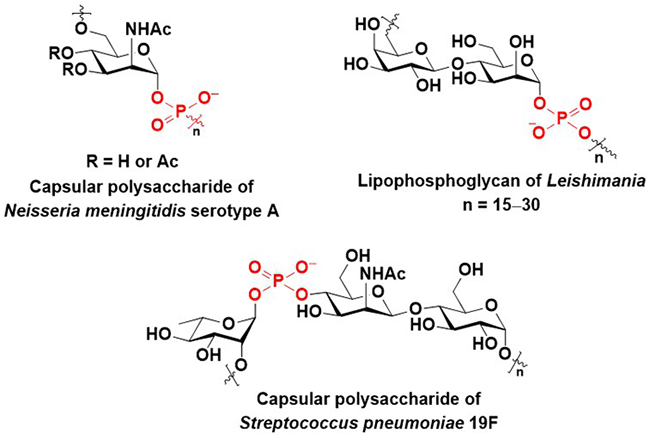
Glycosyl phosphate repeating units are found in the capsular polysaccharides of Neisseria meningitidis1 and Streptococcus pneumoniae2 (Figure 1). Capsular polysaccharides help these pathogens avoid the immune system responses of hosts by masking the highly antigenic constituent of the pathogens3,4. In addition, capsular polysaccharides are recognized as antigens by the immune system of hosts5. The lipophosphoglycans (LPGs) of Leishmania contain characteristic disaccharide 1-phosphate repeating units (Figure 1)6. It has been reported that LPGs play important roles in the survival and infection of Leishmania, and LPGs are recognized as antigens6. Thus, the chemical synthesis of these biomolecules and their chemical analogs can help to elucidate their biological functions and facilitate the development of vaccines and pharmaceuticals. This paper describes the methods developed by our group for the synthesis of glycosyl phosphates using phosphonic acid derivatives.
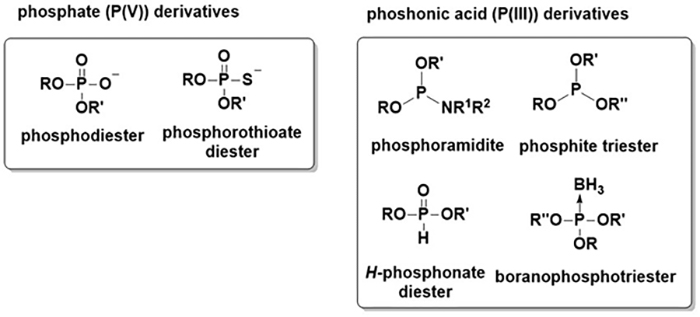
In the human body, phosphorus atoms mostly exist in the form of phosphate (oxidation number of phosphorus: +5)7. Compounds containing a phosphorus atom whose oxidation number is +3 are classified as phosphonic acids. Representative phosphonic acid derivatives are phosphoramidite, phosphite, and H-phosphonate. Boranophosphate, a major research focus of our group, contains a P→B bond and is classified as a phosphonic acid derivative (Figure 2).
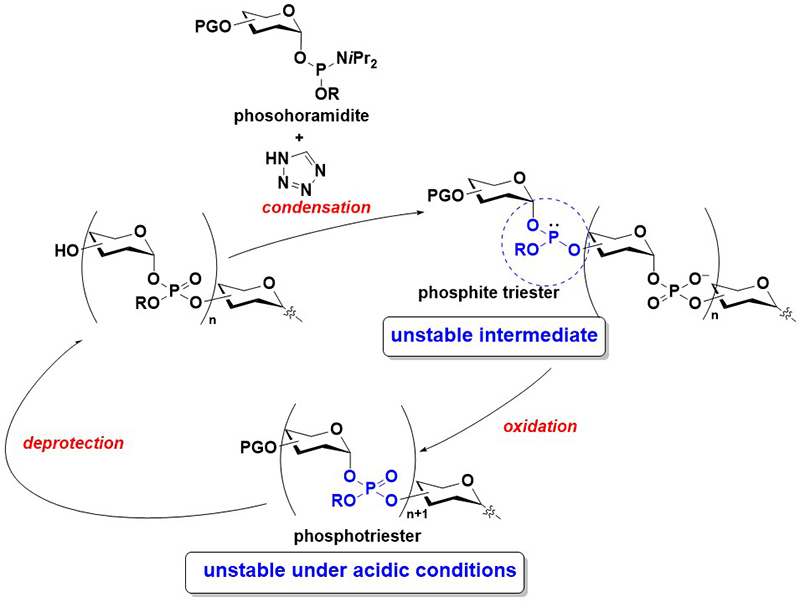
Conventionally, glycosyl phosphate repeating units are mainly synthesized through the H-phosphonate method8. In this method, a glycosyl H-phosphonate monoester is condensed with a hydroxy group using a condensing reagent, such as pivaloyl chloride (PivCl), to form an H-phosphonate diester linkage. Considering that the H-phosphonate diester is unstable, particularly under basic conditions, the H-phosphonate linkage is subsequently oxidized to its phosphodiester counterpart. Thereafter, the deprotection of one of the hydroxy groups and a condensation reaction with another glycosyl H-phosphonate diester allow for further elongation. Although this method affords relatively short oligomers in high yields, challenges remain in the synthesis of long oligomers. First, the oxidation reaction of an H-phosphonate diester to a phosphodiester is mainly conducted using iodine and water in pyridine, and this can lead to the decomposition of an intersugar linkage9,10. Second, the resulting phosphodiester linkage is nucleophilic and can react with a condensing reagent in the subsequent condensation reaction, which may complicate the reaction. Probably due to these problems, the largest number of phosphate repeating units synthesized via the H-phosphonate method is limited to four11.
The phosphoramidite method is the most versatile method for synthesizing oligonucleotides12. The synthesis is generally conducted on a solid support. In this method, a phosphoramidite monomer is condensed with a hydroxy group on the solid support to form a phosphite triester, followed by the oxidation of the phosphite triester to a phosphotriester. Afterward, the removal of a hydroxy protecting group (typically a 4,4′-dimethoxytrityl (DMTr) group) is performed, and the cycles of condensation, oxidation, and deprotection are repeated for the elongation of the oligomer. During the final stage of the synthesis, the deprotection of phosphate moieties and nucleobases, as well as the release from a solid support, are conducted to obtain oligonucleotides. Owing to the high reactivity of the phosphoramidite monomer, this method affords oligonucleotides in good yields. However, to synthesize glycosyl phosphate repeating units, the liability of reaction intermediates must be considered. Specifically, the glycosyl phosphite triester and phosphotriester are unstable, particularly under acidic conditions, and prone to decomposition during synthesis (Figure 3). The most challenging task in the synthesis of glycosyl phosphates via the phosphoramidite method is the prevention of the decomposition of the reaction intermediates.
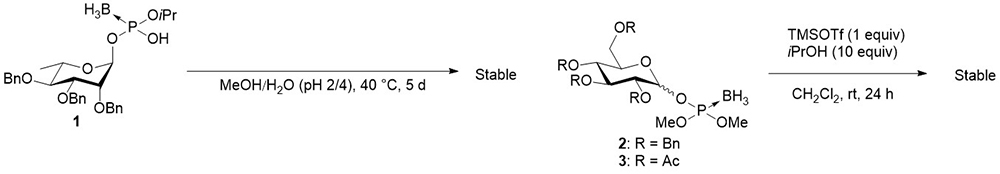
Our group focused on the synthesis using glycosyl boranophosphate as a precursor of glycosyl phosphates. As previously mentioned, a glycosyl boranophosphate is a phosphonic acid derivative, in which one of the non-bridging oxygen atoms of a glycosyl phosphate is replaced by a borano group. Prosperi et al. reported that a glycosyl boranophosphate was intact under acidic conditions in a mixed solvent of MeOH/H2O (pH 2/4) at 40°C for 5 days (Figure 4)13. In a study by Matsumura et al., glycosyl boranophosphates 2 and 3 exhibited no sign of decomposition in the presence of 1 and 10 equivalents of TMSOTf and 2-propanol, respectively, for 24 h (Figure 4)14. Under these conditions, a glycosyl phosphite and glycosyl phosphotriester easily undergo glycosylation. The results showed that glycosyl boranophosphates are more chemically stable than glycosyl phosphites and glycosyl phosphates. A glycosyl boranophosphodiester can be converted into an H-phosphonate diester intermediate, which is a useful precursor of phosphate derivatives, by treatment with a trityl cation15. Thus, it is anticipated that an efficient synthesis of glycosyl phosphate derivatives can be achieved by forming intersugar boranophosphotriester linkages and converting such linkages into phosphate counterparts during the final stage of the synthesis. The results obtained using the strategy are subsequently explained.
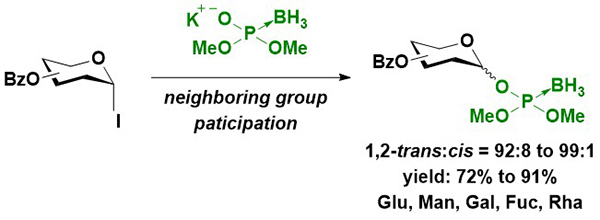
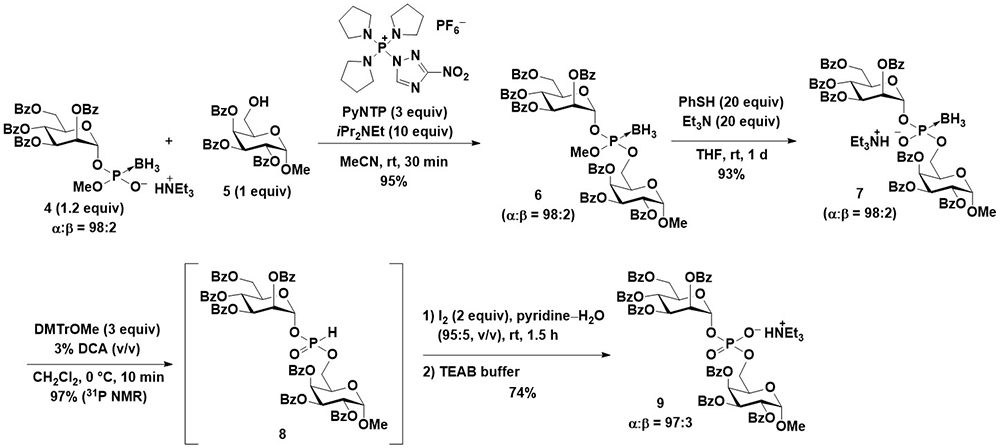
First, the synthesis of glycosyl boranophosphates with the stereocontrol of anomeric linkages was attempted. Imamoto et al. reported the β-selective synthesis of a glucosyl boranophosphate from a glucosyl bromide16. A more reactive glycosyl iodide was used for the reaction, and the reaction of a glycosyl iodide and boranophosphodiester proceeded in an efficient and 1,2-trans selective manner using an acyl-type hydroxy protecting group (Figure 5). The 1,2-trans selectivity was attributed to the participation of the neighboring group, and the major side reaction was the reduction of a dioxocarbenium intermediate to an acetal. Although the use of an aliphatic ester-type hydroxy protecting group promoted the side reaction, the utilization of aromatic ester-type hydroxy protecting groups effectively suppressed the formation of an acetal and improved the yield of the glycosyl boranophosphate. After the glycosyl boranophosphotriester was converted into its diester counterpart 4 by the removal of one of the protecting groups, the resulting boranophosphodiester was condensed with a galactose derivative, 5, bearing a free hydroxy group at the 6-position using a phosphonium-type condensing reagent (PyNTP) developed in our laboratory17. The condensation reaction proceeded efficiently, and a disaccharide derivative 6 bearing intersugar boranophosphotriester linkages was obtained in a high yield. Thereafter, the intersugar linkage was deprotected to a boranophosphodiester (7), converted into an H-phosphonate diester 8 by treatment with a DMTr cation, and oxidized into a phosphodiester 9 using iodine and water in the presence of pyridine. The stereopurity of the disaccharide derivative 9 was practically the same as that of a monosaccharide glycosyl boranophosphate, 4, indicating that the reactions proceeded in a stereospecific manner (Figure 6)18.
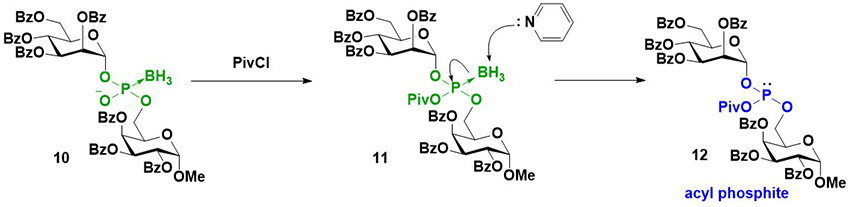
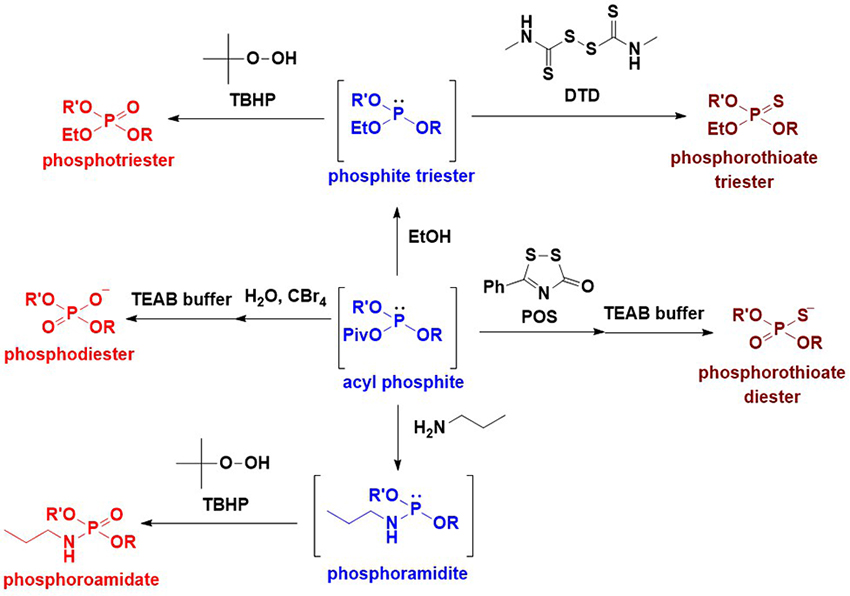
As previously mentioned, an intersugar phosphodiester linkage was effectively constructed using glycosyl boranophosphates. A key reaction in this strategy is the transformation of a glycosyl boranophosphodiester into a phosphodiester or P-modified phosphate derivative. To enhance the utility of the synthesis of glycosyl phosphate derivatives using a glycosyl boranophosphate, a new method for the conversion of a glycosyl boranophosphate into a glycosyl phosphate and P-modified analogs was explored. It has been reported that a P→B bond weakens with an increase in the steric hindrance around a phosphorus atom and a decrease in the electron density of a phosphorus atom19. Thus, it is expected that by introducing a bulky and electron-withdrawing moiety to a boranophosphate, the P→B bond is destabilized and a borano group is easily removed by a nucleophile to afford a phosphite triester-like intermediate. The modification on the phosphorus atom of the intermediate can afford various P-modified glycosyl phosphate derivatives. Based on this concept, we examined the reaction of a glycosyl boranophosphate and an electrophile and discovered that treatment with PivCl in pyridine almost quantitatively furnished an acyl phosphite intermediate (Figure 7). The formation of an acyl phosphite is attributed to the bulky and electron-withdrawing nature of the pivaloyl group, which facilitates deboronation by pyridine. Subsequently, the transformation of a glycosyl boranophosphate into P-modified glycosyl phosphate analogs via an acyl phosphite was investigated. The reaction of a glycosyl boranophosphodiester with PivCl in pyridine in the presence of a sulfurization reagent almost quantitatively afforded a phosphorothioate diester counterpart. The phosphodiester was obtained by the hydrolysis of an acyl phosphite in the presence of CBr4. The reaction of an acyl phosphite intermediate with an amine afforded a phosphoramidite, and oxidation on the phosphorus atom furnished a phoshoramidate. Similarly, a phosphite triester was obtained from an acyl phosphite by treatment with an alcohol, and the oxidation and sulfurization of the phosphite triester afforded a phosphotriester and phosphorothioate triester, respectively (Figure 8)20. The latter was a doubly P-modified analog, whose synthesis is challenging via the existing methods, such as the H-phosphonate and phosphoramidite methods. The results suggest that a glycosyl boranophosphate and acyl phosphite are valuable precursors of glycosyl phosphates and P-modified analogs.
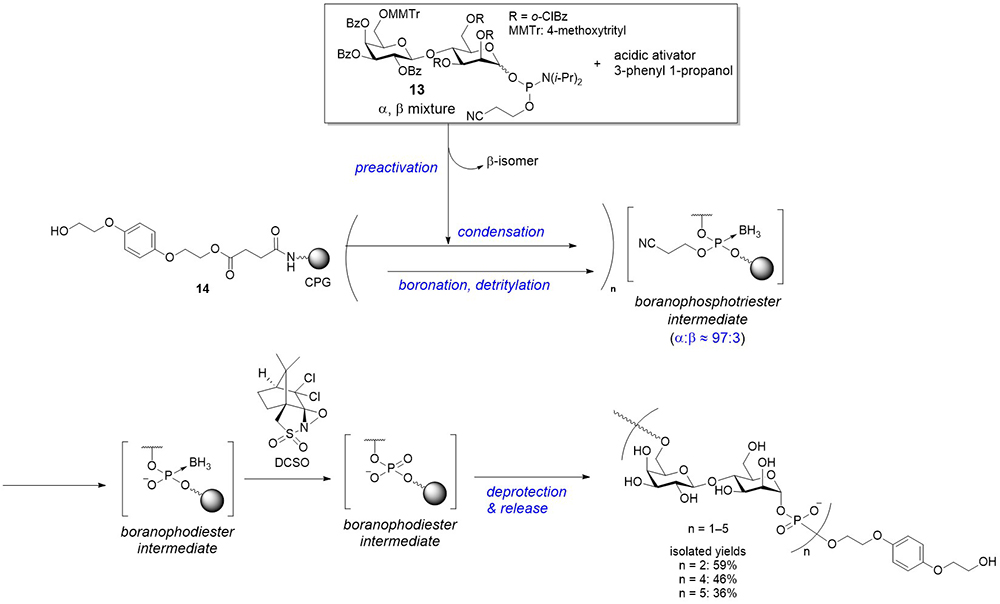
As previously described, a new method for synthesizing glycosyl phosphates and P-modified analogs was developed using a glycosyl boranophosphate as a precursor. Subsequently, the synthesis of an oligo(glycosyl phosphate) using a glycosyl boranophosphate was investigated on a solid support. The [→6)-β-ᴅ-Gal-(1 → 4)-α-ᴅ-Man-(1-P)] disaccharide 1-phosphate repeating unit structure, found in the LPGs of Leishmania6, were selected as a synthetic target, and an appropriately protected disaccharide 1-phosphoramidite (13) was synthesized as a building block for the synthesis (Figure 9). The major challenge of the synthesis of glycosyl phosphates using the phosphoramidite method is the liability of the reaction intermediates. However, an efficient synthesis was expected through the construction of intersugar glycosyl boranophosphotriester linkages and the conversion of the linkages into a phosphodiester at the final stage.
The solid-phase synthesis was conducted as follows. A disaccharide 1-phosphoramidite building block (13; α:β ratio of approximately 90:10) was preactivated with the mixture of an activator, such as 1-phenylimidazolium triflate (PhIMT), and 3-phenyl 1-propanol. This reaction mixture was transferred to a solid-phase synthesis reactor to condense with a hydroxy group on a controlled pore glass (CPG) solid support, 14. The β-isomer of the building block was more reactive than its α-counterpart; thus, the β-isomer was preferentially consumed prior to the reaction on a solid support, and this preactivation protocol effectively improved the α-selectivity21. The resulting phosphite triester was boronated by treatment with BH3•SMe2 to afford a boranophosphotriester, followed by the removal of a 4-methoxytrityl (MMTr) group on the 6-position of a galactose residue. The cycles of condensation with the preactivation protocol, boronation, and deprotection were repeated to elongate a saccharide chain. The key reaction of this strategy was the conversion of boranophosphate moieties into their phosphodiester counterparts. It was serendipitously found that the boranophosphodiester was quantitatively converted into a phosphodiester by treatment with an oxaziridine derivative ((+)-(8,8-dichlorocamphorylsulfonyl)-oxaziridine; DCSO), although it required a relatively long reaction time (Figure 9). Through this strategy, molecules bearing up to five disaccharide 1-phosphate repeating units were obtained22, which is the longest chemically synthesized oligomer with a controlled chain length.
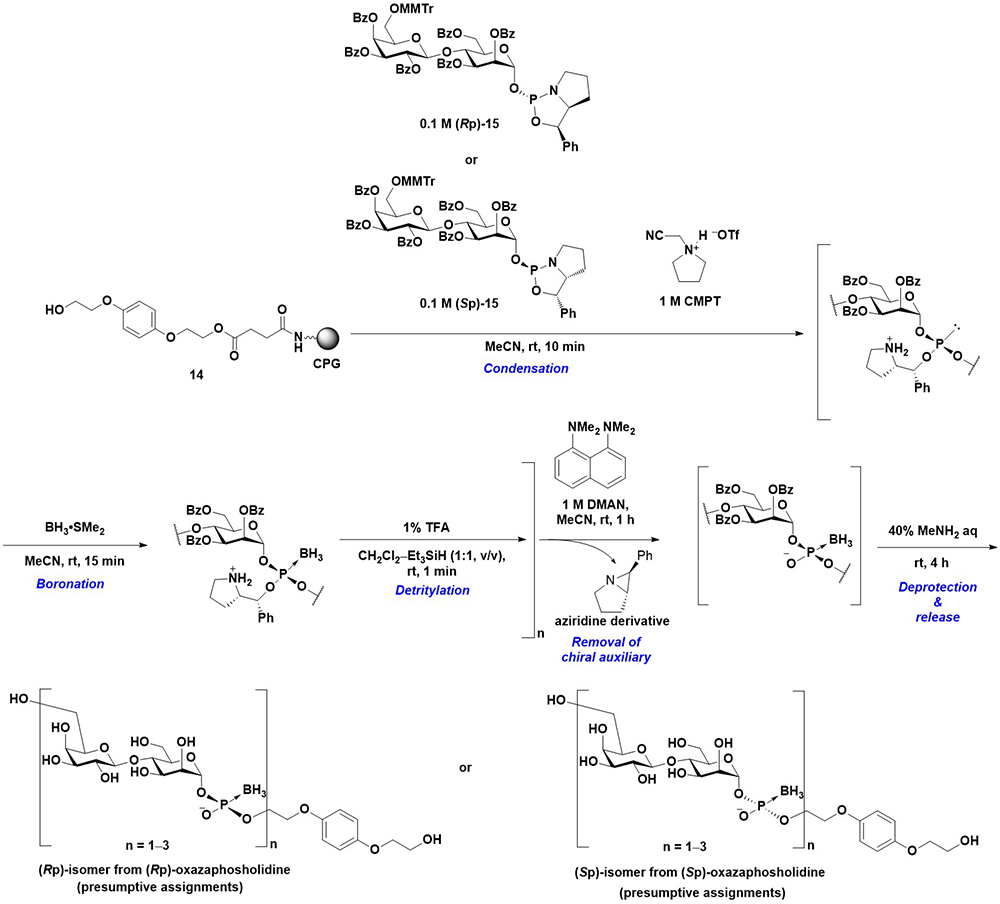
In the preceding section, the synthesis of oligo(glycosyl phosphates) on a solid support using a glycosyl boranophosphate as a stable intermediate is described. P-Modified glycosyl phosphate derivatives, including glycosyl boranophosphates, are potential targets for drug discovery and probes for the elucidation of saccharide recognition. For example, Nikolaev et al. synthesized P-modified [β-ᴅ-Gal-(1→4)-α-ᴅ-Man-(1-P)] structures, including boranophosphate, phosphorothioate, and methylphosphonate derivatives23. It has been reported that the disaccharide 1-phosphate structure is recognized by an elongating mannosyl phosphate transferase (eMPT) and that a mannosyl phosphate moiety is attached to the 6-position of the galactose residue. Nikolaev et al. evaluated the acceptor activity of P-modified analogs and observed that the phosphorothioate and boranophosphate exhibited approximately 75% and 50% acceptor activities, respectively, compared with their phosphate counterpart. However, the methylphosphonate was not an acceptor substrate for eMPT23. The results suggest that phosphorus modification is highly important for recognition by eMPT. In addition, since the phosphorothioate and boranophosphate used in the experiment were considered diastereomixtures, the use of stereocontrolled P-modified analogs would provide insights into the recognition of eMPT. Thus, the stereoconrolled synthesis of P-modified glycosyl phosphate was investigated on a solid support. To control the stereochemistry of phosphorus atoms, an oxazaphospholidine method was employed for the synthesis. An oxazaphospholidine is a cyclic phosphoramidite derivative bearing a chiral auxiliary24. Using a non-nucleophilic acidic activator, the condensation reaction of an oxazaphospholidine and a hydroxy group proceeded in a stereospecific manner to yield a stereocontrolled phosphite triester. The modification of the phosphite moiety affords stereocontrolled P-modified phosphonic and phosphate analogs.
First, a disaccharide 1-oxazaphospholidine derivative (15) was synthesized as a building block with a high anomeric and phosphorus stereopurity. Thereafter, the stereocontrolled synthesis of a glycosyl boranophosphate was investigated. The oxazaphospholidine building block was condensed with a hydroxy group on 14 in the presence of 1-(cyanomethyl)pyrrolidinium triflate (CMPT)25 as an acidic activator, and the resulting phosphite triester was boronated to a boranophosphotriester derivative. Thereafter, the MMTr group on the 6-position of the galactose residue was removed. The cycles of condensation, boronation, and the removal of the MMTr group were repeated, followed by the removal of chiral auxiliaries by treatment with 1,8-(dimethylamino)naphthalene (DMAN). Finally, the deprotection of the other hydroxy groups and release from the solid support afforded a glycosyl boranophosphate in a stereoselective manner (Figure 10). The Rp and Sp isomer of glycosyl boranophosphates were assumed to be obtained from Rp and Sp oxazaphospholidine monomers, respectively. The refinements for an efficient synthesis were as follows. (1) The use of DMAN for the removal of chiral auxiliaries was crucial. The chiral auxiliaries were removed as aziridine derivatives with the activation of a pyrrolidine moiety by a nucleophilic base. The utilization of a more nucleophilic base resulted in the decomposition of the product, probably via deboronation. (2) The washing step with EtOH after the boronation reaction was important for improving the efficiency of the following condensation reactions. A residual boronation reagent interfered with the progress of a condensation reaction, and EtOH was effective in removing the boronation reagent26.
Furthermore, the phosphorothioate counterpart was obtained by the sulfurization of the phosphite triester intermediate26. The synthesized stereodefined P-modified glycosyl analogs can function as valuable probes for the elucidation of eMPT recognition.
Efficient syntheses of glycosyl phosphates were conducted using glycosyl boranophosphate as a stable precursor, and an oxaziridine as a key reagent for the conversion of a glycosyl boranophosphodiester to a glycosyl phosphodiester. The conversion reaction of glycosyl boranophosphate into P-modified glycosyl phosphate was developed using an acyl phosphite intermediate. Considering that a glycosyl boranophosphate is an attractive candidate for pharmaceuticals and probes for saccharide recognition, a method for the stereocontrolled synthesis of glycosyl boranophosphates was established via an oxazaphospholidine approach. I hope to apply the described methods for pharmaceutical and vaccine applications.



