
氏名:梶 裕之
東海国立大学機構 名古屋大学 糖鎖生命コア研究所 特任教授
1984年青山学院大学理工学部卒業、1989年理学博士取得、生物化学研究室助手に着任。1993年東京都立大学理学部助手を経て、2007年産業技術総合研究所に入所、糖鎖医工学研究センター・チーム長、創薬基盤研究部門・グループ長、細胞分子工学研究部門・上級主任研究員、生体システムビッグデータ解析オープンイノベーションラボ・副ラボ長に従事し定年退職。研究テーマ前半は構造生物学、後半はグライコプロテオミクス。2022年より現職。
2023年4月文部科学省大規模学術フロンティア促進事業、ヒューマングライコームプロジェクト(HGA)が始動した。このプロジェクトでは科学目標として「拡張セントラルドグマの実像を明確にする」ことが掲げられ、その実現のために4つの施策が計画されている。その最初の一つ、セグメント1のミッションは、「ヒト糖鎖構造情報を、グライコプロテオームレベルで取得」することである。糖鎖は多様で不均一、産生する細胞、タンパク質、さらにはその部位ごとに異なり、細胞状態の変化に応じて変化する、と言われている。このセグメントでは、断片的、個別的に集積された情報からではなく、規格化された手法で系統的に収集された網羅的情報から、その実態に迫る。まずは血清・血漿糖タンパク質の全容解明に挑む。
糖鎖にはいくつかの存在形態があるが、セグメント1の解析対象は、タンパク質に結合している糖鎖である。オミクスレベルの大規模情報をハイスループットに収集するため、質量分析(LC/MS; Liquid chromatography / mass spectrometry)を基盤とするプロテオミクスの手法を利用する。得られる構造の精密度は用いる手法によって決まる。MS単独では、マンノースやガラクトース、グルコースといった同質量の単糖の区別は困難なので、Hex(5)HexNAc(4)Fuc(1)NeuAc(1)のような組成情報を収集する。あるタンパク質のある部位、特にアスパラギン(Asn)残基側鎖にこのような組成の糖鎖が付いていたとすると、多くの糖鎖関連研究者は、生合成過程や糖鎖合成酵素(糖転移酵素)の局在、活性、特異性などから複合型2本鎖構造を想像するであろう。おそらく大筋としては思い浮かべた構造が正しいであろうが、実際は分からない。フコース(Fucose: Fuc)やシアル酸(NeuAc: N-acetyl-neuraminic acid)はどこに付いているのか、シアル酸の結合はα2,3か2,6か、Galの結合はβ1,4か1,3か、本当に2本鎖なのか、LacdiNAc (N,N’-diacetyl-lactosediamine)をもつハイブリッド型であることもある。個別解析では多角的な詳細解析から正しい構造を見出すこともできるが、大規模解析では組成と一部の構造モチーフまでが収集される。それでも糖鎖の構造制御機構や機能解析の端緒を見出すために役立つ情報を提供できると考えている。
これまで糖タンパク質の糖鎖は、合成される細胞によって異なる、同じ細胞で作られてもタンパク質ごとに異なる、また、同じタンパク質でも、糖鎖付加部位ごとに異なる、といわれてきた。さらに、産生細胞の状態、たとえば分化状態が変わった場合や、がん化、炎症、栄養状態、刺激などによって変化することが知られている。これらの知見は、注目するタンパク質上の、あるいはタンパク質にこだわらずグライコームの相違、変化として解析された結果を基盤とした情報で、全容あるいは、個々のタンパク質、付加部位上の実態はわからない。本セグメントでは、どのソース(健常人プール血清やHEK293細胞破砕抽出物など)のどのタンパク質のどの部位に、どのような(組成の)糖鎖が存在しているか、それらのおおよその多寡はどうか、できれば、どういった状況でそれらはどう変化するか、などを同じ分析基盤の上で系統的に収集し、糖鎖構造に関する情報基盤を整備して、糖鎖の機能や関連する生命現象の機構を解き明かす研究の一助とすることを目的にする。本項では、ヒトゲノム計画、ならぬ、ヒト糖鎖計画セグメント1のミッション、解析方法の原理、課題、現状、および今後の挑戦を概説する。
セグメント1のミッションは、ヒト糖鎖精密地図の作成である。具体的には、どのタンパク質のどの位置にどんな糖鎖が付いているか、を決定することが目的である。したがって、セグメント1で取り扱う分析試料の形態は糖鎖ではなく糖ペプチドとなり、これらのコアペプチドの配列と糖鎖付加位置、部位ごとの糖鎖組成を3つの階層として網羅的に収集する。では糖鎖の精密地図とは何を指すのか。地図の意味するところに向けて順番に説明する。
糖鎖は結合している部位によって主に2つに大別され、一つはアスパラギン残基の側鎖アミド窒素に結合したN結合型、もう一つはセリン・スレオニン残基の側鎖酸素に結合したO結合型である。セグメント1で扱うのは、N型と、O型糖鎖のうち、最初にGalNAc(N-アセチルガラクトサミン)が結合し、さらにこれが修飾・伸長されたムチン型糖鎖、を優先することにしている。網羅的な解析の測定基盤は質量分析法で、分析の前段に液体クロマトグラフィーを接続した、いわゆるLC/MS法を用いる。試料糖ペプチドでは、ペプチド部分が7〜40残基(分子量で約700~4,500)、糖鎖部分がN型で、トリマンノシルコアのみ(Hex(3)HexNAc(2))から仮に4本鎖、例えばHex(7)HexNAc(6)Fuc(2)NeuAc(4)くらいまでとすると、分子量は900〜3,800で、糖ペプチドの分子量は合わせて1,600から9,300となる。シアル酸の多寡、ペプチドの長さや組成、配列によってイオン化効率が異なり、すべての糖ペプチドを同定することは困難であるが、質量分析におけるサーベイ(MS1)分析のレンジを、m/z 400〜2,000に設定すれば、多くの糖ペプチドは2〜5価のイオンとしてカバーし得る。
では目標設定の観点から、網羅的に解析すべき対象がどのくらい存在しているかを考える。まずは糖鎖修飾されるタンパク質の規模について。N型糖鎖とO型糖鎖は生合成過程が異なり、N型の糖鎖修飾はOST (oligosaccharyltransferase)、O型はppGalNAcT(polypeptide GalNAc transferases) によって開始される。これらの酵素はいずれも小胞体あるいはゴルジ体内腔に存在する。したがって、糖鎖修飾を受けるタンパク質は、分子全体あるいは一部が、これらのオルガネラ内腔に入らなければならない。このいわゆる分泌経路に入るためには、翻訳されたポリペプチドのアミノ末端付近に疎水性残基に富むシグナル配列(小胞体内腔で切り離される)、あるいはシグナルアンカー(切り離されずに膜貫通領域として残る)となる領域、もしくは分子のどこかに膜貫通領域となる疎水域が必要なことが知られている。これらの領域を予測すると、ヒトゲノムのコードするタンパク質のアミノ酸配列(約2.2万)のうち、約7千が分泌あるいは膜貫通タンパク質であると予想される。
では次に糖鎖付加部位数である。N型糖鎖は、リボソームで合成されている新生鎖が小胞体トランスロコン(タンパク質透過チャンネル)を通って、小胞体内に入ってくるところで待ち伏せするOSTによって付加される。OSTは基質タンパク質のアミノ酸配列を認識し、Asn-Xaa-(Ser/Thr)(XaaはPro以外)という配列(コンセンサス配列)が存在すると、これに大きなN型糖鎖前駆体をブロックで (en bloc) 付加する。コンセンサス配列の3残基目がCysであっても結合されることがある。OSTの基質(配列)特異性は非常に高いと思われるが、それ以外の配列で全くつかない、ということでもないようだ。トランスロコンから出てきたところに付加されるので、コアタンパク質はまだフォールドする前の状態であり、いわゆるタンパク質のフォールディングやサブユニット間の相互作用に起因する立体障害によって、糖鎖の付加率が大きく下がる事は無いと想像される。だからと言って付加率はどこも100%ではない。付加率に関わる因子の一つに、コンセンサス配列間の近さがある。最短はオーバーラップしている場合で、NNSSやNNTSなどである。以前、糖鎖付加部位の配列と周辺の数残基を大規模に比較した際、このような連続部位の両方に糖鎖が結合した形跡は見られなかった1,2。これが本当だとしたら、糖鎖の立体障害で一方にしか結合させられなかった、あるいは、障害はなかったとしても、膜に埋まった前駆体(ドナー)の供給が間に合わなかった、などが想像されるが証拠はない。タンデムに並んだNXSNXTのような場合、両方についた形跡が見られた。このように一つのペプチドに複数のコンセンサス配列がある場合、一方にしかついていないことが比較的頻繁に見られる。このことは、ドナー基質の供給が間に合わない、という理由はあり得そうだが、常に後から現れるC末側の部位が低頻度、ということでもないので、配列の好み(Preference)で、一方に偏ることも分散することも、両方につくことも起きているように見える。バクテリアOSTの触媒ドメインの結晶構造から、OSTにはThrのメチル基と相互作用する部位があり、OSTはNXSよりNXTとの方が親和性が高い3。同定された付加部位の3残基目としては、SerよりThrの頻度が高い(ポテンシャル配列の総数はほぼ同数であるが、同定数ではNXTがNXSの2倍高かった)。これはNXTの占有率が上がった結果かもしれない。一方、高い占有率で糖鎖が付加されていたにもかかわらず、近くに新たな付加部位が出現したら、元の場所の占有率が著しく下がったケースもあるので、配列だけでなく、別の要因で付加が制御されていることは十分に考えられるが、その機構はまだ不明である。ちなみに、4残基目がProであることも極めて少なかった。SARS-CoV2のスパイクタンパク質について、部位ごとの糖鎖付加を分析した際、各部位の占有率は高かった4。感染された細胞で小胞体は大量のスパイクタンパク質を合成し、その上に忙しなく糖鎖を付加した(祖先型で22箇所ある)と思われるが、占有率の高さからドナー基質が足りなくなる、つけ損なう、というような事はあまりないのかもしれない。
では次にコンセンサス配列であるが糖鎖がつかないケースについては、まず膜トポロジーを考える1。OSTだけでなく、ppGalNAcTも小胞体/ゴルジ体の内腔(Lumen side)にあるので、膜の反対側、すなわちサイトゾル側では当然、糖転移は起こらない。ポテンシャル部位が膜貫通領域内にあっても同様である。アクセプター部位が内腔側にあっても、糖転移酵素は膜タンパク質で、ポテンシャルサイトが膜近くにある場合は、そこに触媒部位が近づけないことも想像に難くない。N型コンセンサス配列を数えると、約7,000配列の潜在的な糖タンパク質には平均で5カ所のコンセンサス配列がある。一部はサイトゾル側に存在するなどで多少数は減るであろうが、約35,000箇所がN型糖鎖付加のポテンシャルサイトと言える。サイト数としてはこの数であるが、実験上では、一つの部位を含むペプチドは消化ミスによって複数存在する上、近傍の残基の修飾(例えばMetの酸化、脱アミド化など)の有無、一つのペプチドイオンが2~4価で存在する、などを考えると、LC/MS 分析で観測されるコアペプチドの質量電荷比(m/z)はおよそ10万点くらいあるのでは、と想像している。我々は予測によるこの候補部位を白地図と捉え、これらのうちどの部位に実存するかを分析し、確定してゆく。できあがった実存リストがコアの精密地図である。地図と呼ぶ理由はもう一つあるがそれは後述する。
では最後に糖鎖組成である。糖鎖の複雑性を述べるとき、ヒトの場合、単糖の種類は主には7種 (Man, Gal, Glc, GalNAc, GlcNAc, Fuc, NeuAc)で、それぞれ3〜4つの結合部位、2種の結合様式(α/β)を持つので、高度に多様な構造をとる、といわれる。しかし、質量分析で検出可能なN型糖鎖の組成は筆者の経験では300種くらいである。結合の部位や向きを考えると、一つの組成でも多数の構造が存在するので、構造の分解能はまだまだ低いが、組成情報だけでも不均一性の実態を垣間見ることができるであろう。
HGAセグメント1の最初の分析対象は、血清および血漿で、比較的低侵襲で収集しやすく、疾患や健康状態を測る(診断のための)生体試料として多くのリソースや情報が集積されている。糖鎖としてははじめにN型糖鎖に重点を置いて、糖タンパク質コアペプチドの配列、糖鎖付加部位、および各部位上の糖鎖組成、の三つの階層の情報を収集する。糖タンパク質と付加部位の同定は、筆者が20年ほど前に開発した安定同位体で糖鎖付加部位を標識し、LC/MS法で取得したMS2スペクトルを元に、データベース検索する方法 (Isotope-coded Glycosylation site-specific tagging; IGOT法) で行う(図 1)5。各部位の糖鎖組成は、糖ペプチドの状態でイオン化し、ペプチド部分と糖鎖部分の両方を適度に断片化して、その断片のスペクトル(MS2) から両者を同定する。しかし、糖ペプチド同定には、イオン化、断片化の問題に加えて、帰属を誤らせる構造特性が複数あるため、今も困難であり、同定ソフトには技術的な課題が残されている。ここでは、HGAにおけるデータ収集の方法と課題、HGAで取り組む挑戦を概説する。
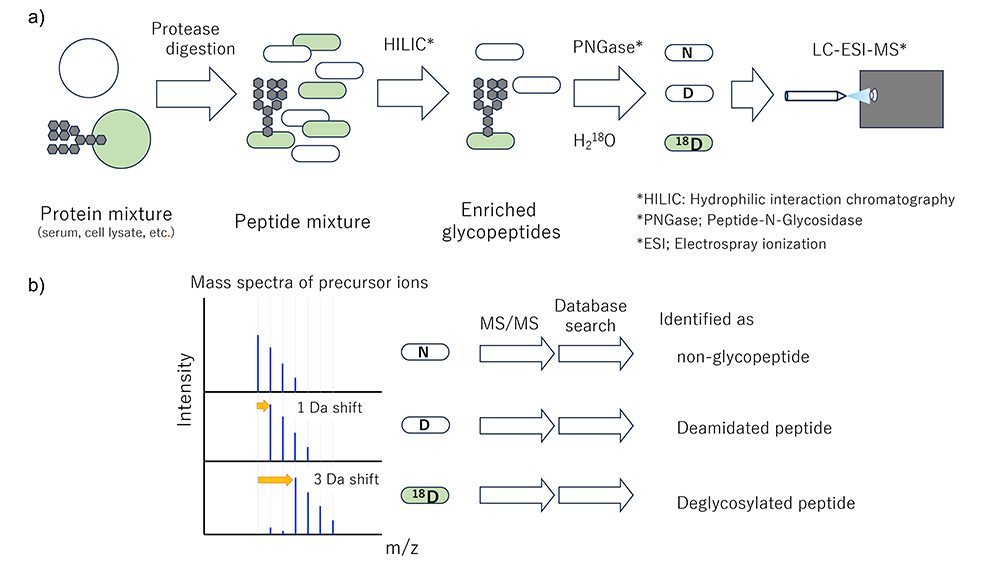
分析試料の形態は糖ペプチドであるが、その検出、同定の感度は、非修飾ペプチドと比較して平均的に低く、筆者の経験(感覚)では2桁以上低い。その要因は後述する。プロテオーム解析においても、消化ペプチドのイオン化効率は長さや組成によって異なり、容易にイオン化するものも、そうでないものもある。しかしタンパク質の同定や定量では、イオン化効率のよい、いわば代表選手を利用すればよい。一方、糖ペプチドは、コアペプチドが必ずしも優秀選手とは限らないため、同定効率は平均して低い。したがって、糖ペプチドを効率的に同定したい場合は、タンパク質消化物から糖ペプチドを捕集するなどして、非糖ペプチドを高度に除くことが必要である。糖鎖の種類に応じた選択的捕集をしたい場合は、レクチンや抗体を用いることなどで達成でき、偏りなく捕集したいときは、親水性相互作用クロマトグラフィーが簡便で、筆者はポリアクリルアミドをリガンドとするアミド80カラム(東ソー)を用いている。セグメント2では、大規模なコホート研究を実施するので、試料収集や糖ペプチド調製は決まった手順で実施され、かつ多数の処理に対応するため、自動化が進められている。セグメント間でプロトコルの大きな乖離の無いよう、確認と調整が図られている。
捕集した糖ペプチドの同定と並行あるいは先行して、糖鎖を切り離したコアペプチドを同定し、タンパク質と糖鎖付加部位をリスト化する(図 1)。糖鎖はイオン化効率を下げ、存在量を下げるので、これを切除すると、プロテオミクスには及ばないが、検出感度が上がる。白地図との対応付けはまず部位同定から始める。糖ペプチドのエンリッチで非糖ペプチドを完全に除くことはできないので、糖鎖切除処理後に同定したペプチドに、付加部位があり、そこに糖鎖付加の痕跡が残っていることが望ましい。コンセンサス配列に付加されたN型糖鎖をPNGaseで切除すると、付加部位のN (Asn)はD (Asp)に変換され、質量が1増えるので、これを痕跡とすることができる。しかし、生体内外では脱アミド化(NからDへの変化)は頻繁に生じるので、痕跡との区別が困難になる。そこで、進行の遅い脱アミド化と進行の速い酵素的糖鎖切除の速度差を利用して区別する方法を考案した。溶媒に18Oで標識した水を使い、この中で糖ペプチドの糖鎖切除を行うと、溶媒の酸素は糖鎖付加部位に選択的に取り込まれ、重くなる(図 1)。筆者らはまずこの方法で、付加部位の網羅的リスト化を試みる。
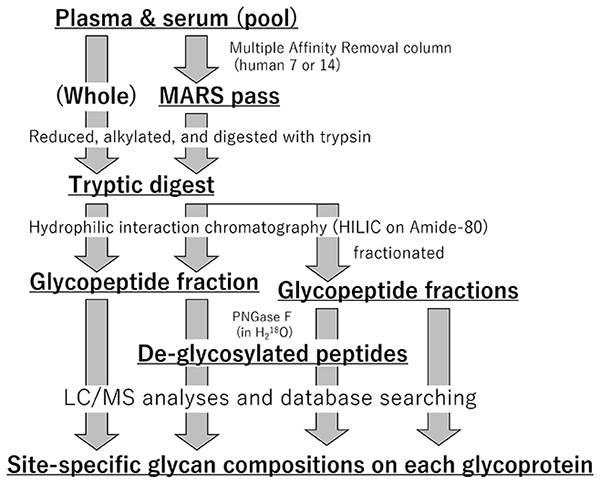
血液試料を扱う上での困難さは、周知の通り、非糖タンパク質であるアルブミンや少数の主要タンパク質の存在である。これらを除去しないものとしたもの両方を用意して糖ペプチドを捕集し、部位の同定および部位ごとの糖鎖解析に用いる(図 2)。データ依存的取得でMS2スペクトルを得る場合には、分析を繰り返すことで同定数が増やせることが知られており、加えて、流速、カラム、グラジェント時間などを効率化して、大規模化を図る。並行して、糖ペプチド試料を分画して複雑性を低下させ、並列的に分析することで、分析深度を深めていく。例えば、そのままの血清等と比較して、初めに主要タンパク質をいくつか除去して、9割のタンパク質が除去されたとすると、検出深度は一桁下がったと考えられる。このポピュレーションを性質の異なる10個に再度分画し、同量ずつ分析すると、深度はもう一桁下げられるであろう。プロジェクト初期に、1,000糖タンパク質について糖鎖付加部位情報の取得を目指す。
糖ペプチドの同定の困難さは多段階にわたっている。イオン化効率の低さ、分子種の分散に加え、壊しにくさ、誤った解釈(帰属)を起こしやすい構造的特徴などが信頼できる同定を阻んでいる。
イオン化効率の低さは特にN型糖鎖のような大きな修飾基が付くことに加え、糖鎖の非還元末端に負電荷を持ったシアル酸が複数付くことなどによる。また検出感度低下の要因の一つは、糖鎖の不均一性による。あるタンパク質が10 fmol存在し、プロテアーゼ消化がうまくできると、各ペプチドは10 fmolずつ生じることになるが、糖鎖付加部位のペプチドに10種の重さの違う糖鎖が付くと、各糖ペプチドの存在量は平均で1 fmolとなる。複合型糖鎖の場合、10種を遥かに超えた数の糖鎖が付加され、分散による感度低下は避けられない。さらに、壊しにくさ、が問題である。MSでの同定基盤は、断片化に依存する。ペプチド同定はペプチド結合で切断された一群の断片イオン(b/yイオン)が生じることで可能になる。糖ペプチドはアミノ酸のオリゴマーと単糖のオリゴマーが結合したもので、ペプチドを形成するペプチド結合より、糖鎖を形成するグリコシド結合の方が弱く、壊れやすいため、弱い(単発的な)衝突誘起解離法では、糖鎖部分が壊れるだけで、ペプチドは壊れず、結果として同定できない。最近は、繰り返し衝突を起こさせる高エネルギー衝突誘起解離法(Higher energy collision-induced dissociation; HCD)によって、ペプチド部分のフラグメントイオンも生成できるようになり、MS2で糖ペプチドの同定が比較的容易になった。糖ペプチドの配列や糖鎖構造は様々で、一つの条件で一様に断片化することはできないので、壊すエネルギーを複数用意し、得たスペクトルを足し合わせてMS2スペクトルを構築することで、多様な糖ペプチドに適用できるようになっている。典型的な糖ペプチドのMS2スペクトルを図 3に示す。弱いエネルギーでは糖鎖のグリコシド結合が切れて、ペプチドとペプチドから近い糖が残った形のイオン(Yシリーズのイオン)が観測され、N型の場合はトリマンノシルコア構造があるので、ペプチド+GlcNAc+GlcNAc+Man+Man+Manというような一連のシグナルが見られる。重さでGlcNAcなどと決めることはできないので、HexNAcと書くのが正しいのであろうが、生合成経路を考えると上記のようになる。このシリーズで有用なのが、ペプチド+HexNAc+dHex、と、ペプチド+HexNAc(3)+Hex(1)のようなイオンの存在で、これらのシグナルはそれぞれ、コアフコースおよびバイセクトGlcNAcの存在を示唆する。Yシリーズのイオンで、一番大きく観測されるのは、ペプチドにGlcNAcが一つ残ったY1イオンで、HexNAcの質量差でY0-Y1-Y2イオンが観測されると、Y0、すなわちペプチドの質量がわかるので、プロテオミクスにおけるMS/MS-イオン検索法が適用でき、ペプチドが断片化できて生じたb/yイオンと、このY0の質量からペプチド同定が可能になる。
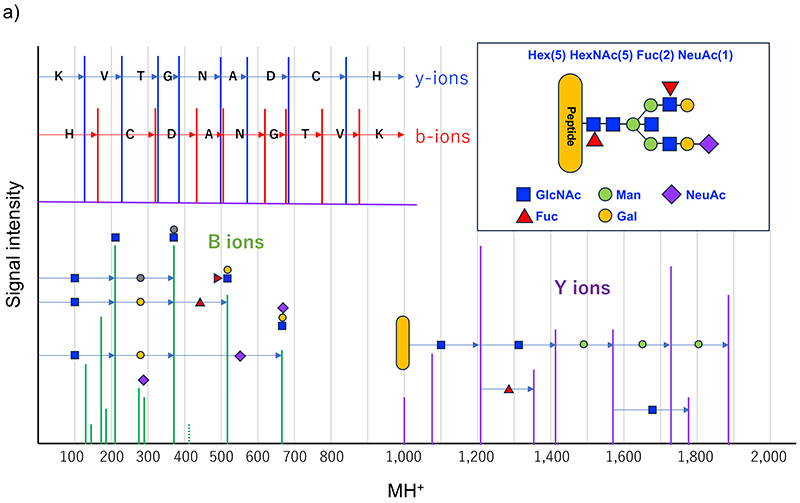
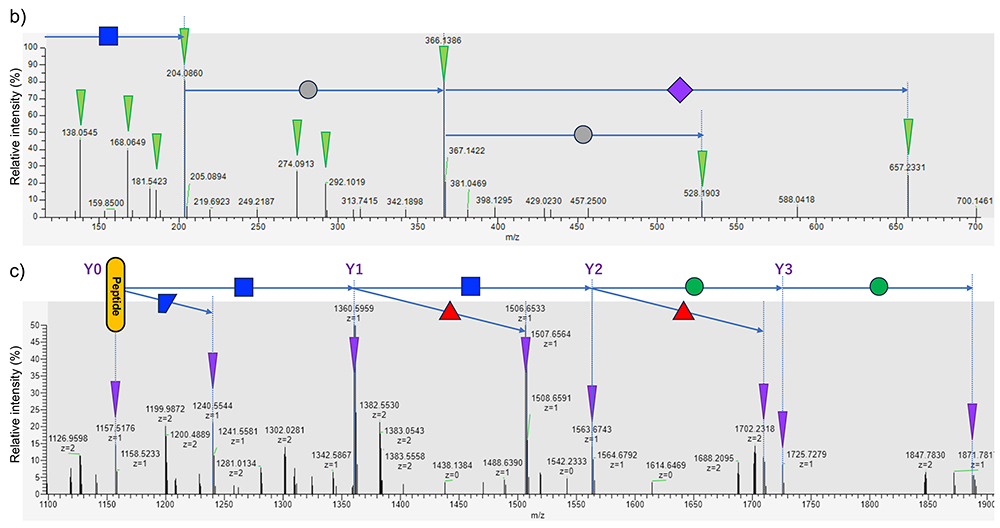
グリコシド結合が一つ、あるいは幾つか切れて生じた糖鎖の断片イオンは、糖鎖の存在や構造的特徴を示唆し、診断イオンとも呼ばれている。HexNAcの存在は、m/z 138, 144, 168, 186, 204などのシグナルで強く示唆される。Hexはイオン化しにくいので弱いが、Gal-GlcNAc-のような構造はm/z 366のシグナルで示唆される。他に、N-アセチルノイラミン酸(NeuAc)に由来するイオン(m/z 292, 274)やNeuGc由来のイオン(m/z 308, 290)はシアル酸の区別をすることに役立つ。単糖だけでなく、よく見られるNeuAc-Hex-HexNAc-(m/z657)のほか、Hex(dHex)-HexNAc (Lewisx/y; m/z512)やHexNAc(2)(LacdiNAc; m/z407)などは含有する構造モチーフを示唆し、構造解析の情報源となる。ただし、これらの診断イオンは、混入したプレカーサーイオンに由来する可能性があるので、十分な注意が必要である。
衝突エネルギーが強まるとペプチド結合が切れてb/yイオンが生じ、ペプチド部分の同定につながる。多くのMS2解析ソフトはこれらの特徴を利用して、ペプチド配列と糖鎖組成(および少しのモチーフ情報)を同定しようとするアルゴリズムが組み上げられているが、いつも同定に十分なフラグメントが出るとは限らないので、糖ペプチド同定の確からしさを評価するのは難しい。さらに、糖鎖組成を誤認させる内因的な構造的特徴がある。最もやっかいな問題は酸素原子の所在による。HexとFucの元素組成の差は酸素一つであるが、NeuAcとNeuGcの差も同じである。したがって、糖鎖質量が同じでも、Hex+NeuAc=Fuc+NeuGcなので質量だけでは区別できない。断片イオンで確認したり、端からヒトではNeuGcは存在しない、と仮定するなどで同定や確認は可能であるが、常に証拠がそろっているわけではないので、同定アルゴリズムによって誤同定が生じる。
また、プレカーサーイオンのモノアイソトープ質量を取り違えると、Fuc(2)=NeuAc+1の関係から誤同定が生じる。モノアイソトープシグナルを誤ることは糖ペプチド、とくに長めのペプチドに大きめの糖鎖が付いた糖ペプチドでは、モノアイソトープシグナルが微弱なためまれではない。また多数のフコース、具体的には5つのFucの質量は2*(Hex(1)HexNAc(1))の質量と差が小さく、糖ペプチド質量が大きい場合、区別できなくなることもある。このように、MS2スペクトルから糖ペプチドを帰属するソフトは未だ開発の余地が大きく、ヒトプロテオーム機構(HUPO)の国際共同研究促進活動(Human Glycoproteomics Initiative; HGI)では、パイロット研究を実施し、現在第2期が進められている6。今後の開発に期待がかかる。
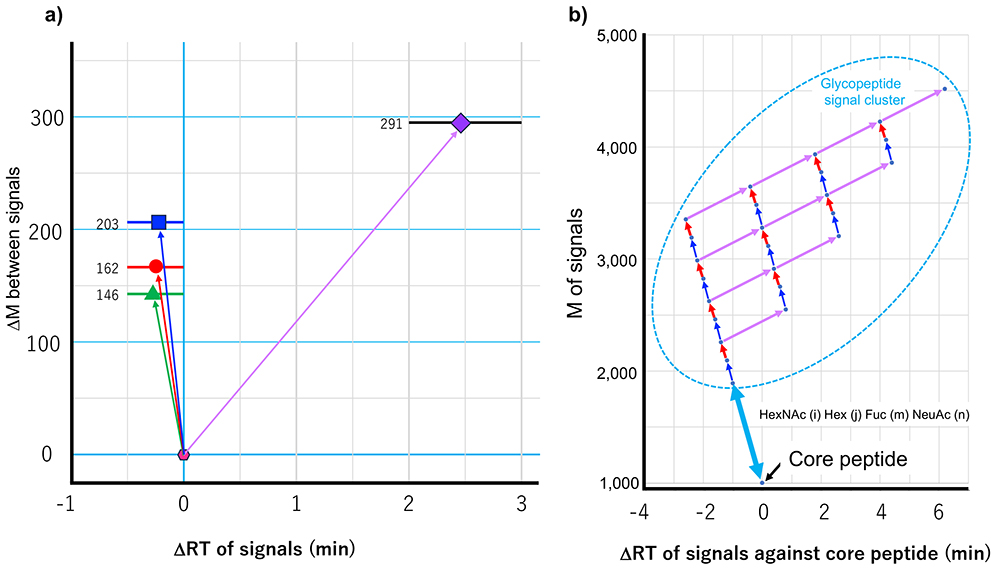
いまや複雑な、例えば細胞抽出物のプロテアーゼ消化物の分析では、わずか100-200ngで1万種以上のタンパク質を同定できる時代であるが、糖タンパク質の場合、サイト同定であっても2千タンパク質を超えるのは難しい。糖鎖が付いていたらなおさらである。本プロジェクトではこの障壁を越えて大規模に同定することを目指すので、高感度化、迅速化は避けて通れない。このための取り組みとして、筆者らが取り組んできたMS2に依存しない高感度同定法の確立、応用と、それによって得られた情報を利用した同定技術(参照同定法)の確立を進めている。MS2に依存しない高感度同定法とは、糖鎖の不均一性と糖ペプチドの溶出特性を利用した同定法である(Glycan heterogeneity-based Relational IDentification of Glycopeptide signals on the Elution profile; Glyco-RIDGEと呼んでいる7,8)(図 4)。これまで述べてきたように、糖鎖が不均一であるために糖ペプチド同定の感度低下が生じているが、この方法は逆にそれを利用する。すなわち、LC/MSで利用する逆相LCにおいて、糖ペプチドは主にペプチド部の疎水性によって分離するため、ペプチド部が同一で糖鎖組成の異なる一群の糖ペプチドは近接した保持時間帯に溶出する。この時、中性糖(Hex, HexNAc, Fuc)が付加すると保持時間は若干早まり、糖鎖が大きくなるほどに保持時間は短くなる。一方、酸性糖(NeuAc)が付くと保持時間は大きくなる。さらに、これら一群の糖ペプチドのシグナル間の質量差は、限られた少数の単糖(質量の観点で4種)のいずれかに限られるため、例えばHex(5)HexNAc(2)とHex(6)HexNAc(2)をもつ糖ペプチドを一対の同一コア糖ペプチドシグナルとして見出すことが可能である。こうして見出されたシグナルのうち、一方がMS2で同定されていれば、他方は同一コアである可能性が高い。一対では偶然の一致によるかもしれないと思われる場合は、3ないし4つでクラスターとして見出すこともできる。検出されたシグナルのクラスターのうち、いずれも同定されていない場合、糖鎖の付いていないペプチドの質量との比較で関連付けすることができる。糖鎖の付いていないペプチドの存在(質量)や保持時間は上述のIGOTサイトマッピングで高感度に検出されている可能性が高いので、これを利用する。質量差と保持時間の関連を基盤にMS2情報無しで同定することに抵抗を感じる研究者もいるであろう。当然、提示する側としても誤った情報(同定結果)を流布することは望んでいない。プロテオミクスの定量分析では、データ依存取得法で得たデータを元に定量する際、一方で同定されていなかったとしても、保持時間と質量からシグナルの同一性を判断してデータ欠損を防ぐ、match-between-runsという手法を利用することが受け入れられている9。質量と質量差を同じように扱うことには賛同できないかもしれないが、信頼度(支持する証拠がどの程度そろっているか)を提示しつつ可能性を提供することには意義があると考えている。研究の糸口を得る一つのツールとして考えて頂ければ幸いである。
プロテオミクスでは、データ非依存的MS2取得(DIA)が同定数の爆発的な増加を導いた。当初はあらかじめ同定に至ったスペクトルとのマッチが必要であったが、今ではペプチドの溶出位置は予測され、プレカーサー及びフラグメントイオンの計算質量との一致度で同定できる。同様に糖ペプチドも溶出位置を予測する事はできるが、前述のように糖ペプチドは糖鎖が違っても近接して溶出され、HCDで生じる断片もほぼ変わらないため、プレカーサーとフラグメントイオンを溶出プロファイルから紐づけることが困難となり、データ非依存であってもMS2に依存していたら同定の高感度化は難しいかもしれない。しかし、MS装置はどんどん高感度、高速化しているので、数年後には容易に同定できているかもしれない、などと楽観的に考えたりもする。一方、糖鎖は細胞ごとに異なる、ので、組織ごとの分析では由来が曖昧になる。突き詰めるとシングルセルでの観測が必要なのであろう。本研究は前もって参照可能な情報基盤を深掘りして整備し、今後の分析に役立てるための取り組みである。エドマン分解でペプチド配列を決定して同定したり、目的タンパク質のcDNAクローンを何年もかけてクローン化して、マキサム-ギルバート法やサンガー法で塩基配列を決定したりしなくても済むようになったように、糖ペプチドの容易な同定基盤を確立したい。当然ながらヒト糖鎖には、このアプローチでは俎上に載らない種類や構造の糖鎖もたくさんあり、例えばシアル酸が連結したオリゴ/ポリシアル酸は検出が困難であったり、そもそも我々が用いる試料調製条件では壊れてしまったりするので、他のアプローチでの解明が進行されている。さらに、最初に述べたように、このアプローチでは糖鎖の組成までしか分からないが、単糖の種類、結合の位置や向きの情報の部位特異的取得についても計画されている。最後にお願いですが、解析ツール作りに研究的新規性はない、とインフォマティシャンには敬遠されがちであるが、これに一緒に取り組んでくれる情報科学者の協力を望んでいる。



