
Hiroyuki Kaji
Designated Professor
Institute for Glyco-core Research, Nagoya University, Tokai National Higher Education and Research System
Hiroyuki Kaji graduated from the College of Science and Engineering, Aoyama Gakuin University in 1984, got a doctoral degree in science, and took up his post as an assistant professor in the University in 1989. He was appointed assistant professor in the Faculty of Science, Tokyo Metropolitan University, in 1993. In 2007, he joined National Institute of Advanced Industrial Science and Technology (AIST), where he served as Team Leader at the Research Center for Medical Glycoscience, as Group Leader at the Biotechnology Research Institute for Drug Discovery, as Chief Senior Researcher at the Cellular and Molecular Biotechnology Research Institute, and as Deputy Director at the Computational Bio Big Data Open Innovation Laboratory, until mandatory retirement at the stipulated age. His research focused on structural biology and then on glycoproteomics. He has been in his current position since 2022.
In April 2023, the Human Glycome Atlas (HGA) Project was started within the framework of the Large-Scale Academic Frontier Promotion Project sponsored by the Ministry of Education, Culture, Sports, Science and Technology of Japan. The HGA Project advocates for the scientific goal of “Clarifying the Truth of the Extended Central Dogma” and has four strategic segments planned to attain this goal. First, the mission of Segment 1 is to “acquire information on human glycan structures at the glycoproteome level”. Glycans are diverse and heterogeneous, and vary depending on producing cells, protein kinds, and glycosylation sites; furthermore, they are described as alterable with change in cellular status. In this segment, the actual status of glycans is explored based on comprehensive information that has been systematically acquired using a standardized approach, rather than piecemeal information acquired fragmentally and compiled individually. First, efforts are underway to elucidate the whole picture of serum and plasma glycoproteins.
Abbreviations: Hex; hexose, HexNAc; N-acetylhexosamine, Fuc: fucose, Man; mannose, Gal; galactose, Glc; glucose, GalNAc; N-acetylgalactosamine, GlcNAc; N-acetylglucosamine, NeuAc; N-acetylneuraminic acid, NeuGc; N-glycolylneuraminic acid, LacdiNAc; N,N’-diacetyl-lactosediamineGlycans come in various forms. The focus of analysis in Segment 1 is protein-bound glycans. To collect a large amount of information at the omics level in high throughput applications, a proteomics approach based on liquid chromatography / mass spectrometry (LC/MS) is utilized. The accuracy of the structural determination varies depending on the approach used. Using MS alone, it is difficult to distinguish among monosaccharides with identical mass, such as mannose, galactose, and glucose. Hence, compositional information such as Hex(5)HexNAc(4)Fuc(1)NeuAc(1) is collected. If we assume that a glycan of such a composition is attached to a site, particularly the asparagine (Asn) residue side chain of a protein, many glycan researchers will imagine a complex type bi-antennary structure from the localization, activity, and specificity of glycan-synthesizing enzymes (glycosyltransferase), the biosynthetic process, and other factors. Although the imagined structure is probably right, the truth is unknown. For example, it is not unclear: Where do fucose and sialic acid (NeuAc) attach? Is the binding mode of sialic acid α2,3 or α2,6? Is the mode of Gal β1,4 or β1,3? Is the structure bi-antennary complex type or a hybrid with LacdiNAc (N,N’-diacetyl-lactosediamine)? Individual analysis is capable of finding the accurate structure by multifaceted approach, whereas in large-scale analysis, compositional data, including some structural motifs, are also be collected. Even so, the author believes we can provide information that should help finding clues to the glycan structure control mechanism and functionality.
Thus far, glycans on protein have been described as differing depending on the cells where they are synthesized, as varying among different proteins even in the same cell, and as varying among different glycosylation sites in the same protein. Furthermore, they are known to change depending on the cell status, e.g., alteration of the differentiation state, carcinogenesis, inflammation, nutrition status, and stimulation. However, these discoveries are based on the results of glycome analyses of specific protein of interest or protein mixtures, the actual state of site-specific glycosylation on each protein is poorly understood. The purpose of this segment is to systematically collect information on glycoproteins, glycosylation sites, and site-specific glycan structures from human-derived sources (e.g., pooled healthy human sera, other body fluids, lysate of HEK293 cells, and tissues) using the same analytical basis. If possible, relative abundance of each glycoforms and how glycans change in different situations are planned to obtain and provide to clarifying the roles of glycans and the mechanism of glycan-relating biological phenomena. This section outlines the mission of Segment 1 in the Human Glycan Atlas Project, as well as the principles, current status, problems, and prospects for the analytical procedures to be used.
The mission of Segment 1 is to generate a site-specific glycan map of all human glycoproteins. Specifically, efforts are underway to determine which glycan is attached to which site in which protein. Therefore, the analytical samples to be handled in Segment 1 are not glycans but glycopeptides, and the data comprehensively collected in three strata on these core peptides will include: sequence, glycosylation site, and site-specific glycan composition. Then, what is meant by the site-specific glycan map? The steps taken to answer this question are described below.
Glycans can be roughly divided into two types depending on binding site: N-linked glycans, whose binding site is nitrogen of asparagine side chain and O-linked glycans, who bound to oxygen of serine / threonine side chain. In segment 1, N-glycans will be analyzed at first, and then O-glycans, especially mucin type (GalNAc-based and elongated forms) are followed. Measurements in the comprehensive analysis are based on mass analysis by liquid chromatography-mass spectroscopy (LC/MS), which involves the use of liquid chromatography in the first half of the analysis. If we assume that the sample glycopeptides have a peptide moiety of 7 to 40 residues (molecular weight approximately 700–4,500) and an N-linked glycan moiety consisting of a trimannosyl core alone (Hex[3]HexNAc[2]) to tetra-antennary glycans, e.g., Hex(7)HexNAc(6)Fuc(2)NeuAc(4), then the molecular weight of the glycan will be 900 to 3,800 and the total molecular weight of the glycopeptides will be 1,600 to 9,300. Because ionization efficiency varies depending on the abundance of sialic acid and the length, composition, and sequence of the peptide, it is difficult to identify all glycopeptides; however, many glycopeptides could be detected as divalent to pentavalent ions by setting the mass range of survey (MS1) window to m/z 400 to 2,000. Now, in order to set goals, let's consider the size of the analysis target that will be comprehensively analyzed.
First, we will consider the size (number) of proteins that are glycosylated. N- and O-glycans are produced by different biosynthetic processes; N-linked glycan is attached by oligosaccharyltransferase (OST) en bloc at first step; and O-glycan modification is started from a binding of GalNAc catalyzed by one of polypeptide GalNAc transferases (ppGalNAcTs). All these enzymes are present in the lumenal space of endoplasmic reticulum (ER) or Golgi apparatus. Therefore, to undergo glycan modification, the protein molecule should entirely or partially enter the lumen of these organelles. It is known that entry into this secretory pathway requires the presence of a signal sequence rich in hydrophobic residues (a sequence to be cut off in the ER lumen), a signal anchor domain (a sequence not cleaved off and remaining as a transmembrane domain) near the amino terminus of the translated polypeptide, or a hydrophobic region functioning as a transmembrane domain anywhere in the molecular structure. Approximately 7,000 of the approximately 22,000 amino acid sequences encoded in the human genome are likely to encode secretory or transmembrane proteins.
The next topic is the number of glycosylation sites. For N-glycoproteins, nascent chains synthesized in the ribosome are introduced into lumen side space of ER through translocon (a protein translocation channel) and then N-glycan is added by OST, which is waiting near the translocon exit. Recognizing the amino acid sequence of the substrate protein, OST adds a large N-glycan precursor en bloc to the consensus sequence Asn-Xaa-(Ser/Thr) (where Xaa is any amino acid except Pro) if present. Glycan binding can occur, even if Cys is the third residue in the consensus sequence. Although the specificity of OST for this substrate (sequence) appears to be extremely high, it is not likely that there is no glycan binding to any other sequences. Glycans are attached to nascent proteins near the exit of the translocon pore. In this case, the core protein is still in its unfolded state, so steric hindrance due to protein folding or subunit interactions is unlikely to significantly reduce the glycosylation rate. Even so, the glycosylation rate is not 100% everywhere.
One factor related to glycosylation rate is the proximity of consensus sequences. The highest proximity is seen in overlapping sequences, such as NNSS and NNTS. When we previously compared glycosylation site sequences and several residues around them on a large scale, we found no trace of glycan binding to both of such adjoining sites 1,2. If this is true, it is likely that glycan binding occurred on one side only due to steric hindrance by the glycan, or, without steric hindrance, due to the momentary inaccessibility of the precursor (donor) embedded in the membrane; however, no evidence is available. In the case of the tandemly arranged motif like NXSNXT, a trace of glycan binding to both sites was found. In the presence of multiple consensus sequences for a single peptide as described above, glycan binding to one site only is relatively common. This may be explained by a temporary shortage of donor substrate (not arriving in time). However, the frequency of glycosylation is not always lower at the C-terminal site, so glycosylation appears to occur at either or both, depending on the preference of the OST. From the crystalline structure of the catalytic domain of bacterial OST, there is a site for interaction with the methyl group in Thr; the OST exhibits higher affinity for NXT than for NXS3. Regarding the third residue in the identified glycosylation site, Thr is more common than Ser. In terms of potential sequences, the number of NXS and NXT was similar, but in the number of identified sites, NXT had approximately twice as many sites as NXS. This may reflect an increased occupancy of NXT. On the other hand, in some cases, the occupancy at the original site decreased remarkably when a new glycosylation site emerged nearby despite glycosylation occurring at a high rate; therefore, it is very likely that the addition is controlled not only by the sequence, but also by other factors; however, the mechanism remains unknown. Additionally, the fourth residue is very rarely Pro. When analyzing SARS-CoV2 spiking proteins for site-specific glycan additions, each site has a high occupancy4. In the ER of infected cells, a large amount of spike protein is synthesized and seems to be added glycans busily (22 sites in the ancestral type); however, due to the high occupancy, lack of donor substrate or addition failure seems unlikely.
Next, in cases where no glycan attaches to the consensus sequence, membrane topology should be considered first1. Because not only OST but also ppGalNAcTs are present in the lumen side of the ER and Golgi, glycosylation should not occur on the opposite side of the membrane, i.e., the cytosol side. This is true in cases where the potential site is located in the transmembrane domain. It is readily imaginable that even if the acceptor site were located on the lumen side, the catalytic site of glycosyltransferases would be unable to approach the potential site close to the membrane of membrane protein since glycosyltransferase is also a membrane protein. When counting N-glycosylation consensus sequences, approximately 7,000 sequences of potential glycoproteins each have five consensus sequences on average. Hence, approximately 35,000 sites may be presumed as potential sites for N-glycosylation, although this number may decrease a bit due to the location of some sites on the cytosol side.
This is the expected number of potential sites, but in experiments, multiple peptides containing one site will be generated due to miss cleavage in protein digestion. In addition, considering the presence of partial modification of nearby residues (for example, oxidation of Met, deamidation, etc.) and the fact that one peptide ion has multiple charge states (e.g., z=+2~+5), it is estimated that there will be over 100,000 ions (m/z) of the core peptides. We will identify which of these predicted sites (we call them blank maps) are actually glycosylated. The resulting list of actually glycosylated sites is a site-specific map of the core. There is another reason for calling the list a map, which is described in a separate section below.
The final topic is glycan composition. Talking about the complexity of glycans, highly diverse structures are formed from mainly seven monosaccharides in humans (Man, Gal, Glc, GalNAc, GlcNAc, Fuc, and NeuAc), each of which have three or four binding positions (2, 3, 4, 6, and 8) and two binding orientations (α and β). However, in the present author’s experience, mass analysis can only detect up to 300 different N-glycan compositions. Because the single composition can include many glycan structures with different binding position and orientations, the structural resolution remains low; however, composition information alone will give us a peek at the actual status of glycan structure heterogeneity.
In HGA Segment 1, the first subject of analysis is serum and plasma, which are easily obtainable by minimally invasive techniques, are the most frequently analyzed specimens. Consequently, there is a large store of data from serum and plasma samples that have been and can be used for diagnostic purposes. First focusing on N-glycans, information is collected on glycoprotein core peptide sequence, glycosylation site, and site-specific glycan composition. Glycoproteins and glycosylation sites are identified by isotope-coded glycosylation site-specific tagging (IGOT), LC/MS analyses (acquiring tandem mass spectra (MS2)) of the resulting deglycosylated peptide, and database searching for MS2 spectra, which method was developed by the authors some 20 years ago (Figure 1)5. The glycan composition of each site is identified based on the MS2 spectrum of the glycopeptide. At that time, the glycopeptide ion is moderately fragmented at both the peptide and glycan moieties. However, glycopeptides are still difficult to identify because problems with ionization and fragmentation of glycopeptides remain. In addition, since glycans have unique structural properties that may cause identification errors, technical issues in identification software need to be resolved. The data collection methods, issues, and challenges in the HGA Project are summarized below.
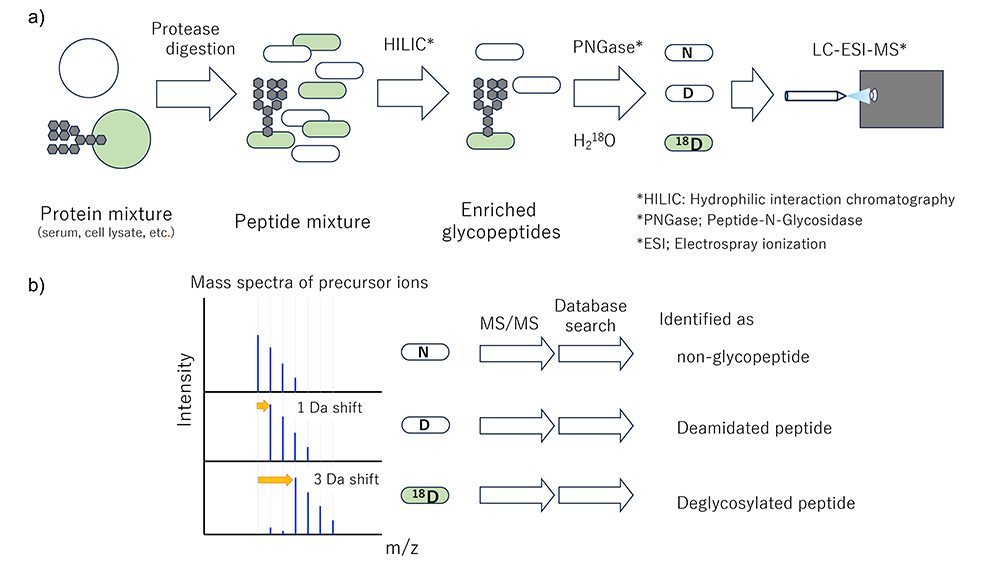
Analytical samples are glycopeptides, and the sensitivity of detection or identification is generally, in the author’s estimation, two orders of magnitude lower for glycopeptides than for non-glycosylated peptides. Relevant factors are described in a separate section below. In proteome analysis, the ionization efficiency for digested peptides varies depending on the length and composition, some of them being easily ionizable, others not. However, protein identification and quantification can be performed with high sensitivity using representative some peptides with higher ionization efficiencies.
On the other hand, in the case of glycopeptides, the core peptide of the glycopeptide does not necessarily have a high ionization efficiency, so the identification efficiency is low on average. Therefore, to identify glycopeptides efficiently, elimination of most non-glycopeptides by, for example, capturing the glycopeptides from the protein digests, is necessary. A lectin or antibody can be used to selectively capture the desired glycopeptides. To perform comprehensive capture, hydrophilic interaction chromatography (HILIC) is conveniently available; the present author uses the Amide 80 column (Tosoh Corporation) with polyacrylamide as the ligand. Segment 2 will involve large-scale cohort studies. Automation of sample collection and glycopeptide preparation is being developed to perform them with established procedures and high throughput. All details are checked and adjusted to avoid large differences in protocols between different segments.
Concomitant with, or in advance of, the identification of the captured glycopeptide, the deglycosylated core peptide was identified and the protein and glycosylation sites were listed (Figure 1). Because glycans lower the ionization efficiency and their heterogeneity reduces the abundance of each glycopeptide, removing glycans improves the detection sensitivity of glycosylation sites, though the sensitivity does not reach the sensitivity of proteomics analysis. Mapping is started for glycosylation site identification. Since it is impossible to completely eliminate non-glycopeptide by glycopeptide enrichment, it is desirable that the peptide identified after deglycosylation have a glycosylation consensus sequence and a clue of glycosylation remaining. When N-glycan is removed with PNGase, glycosylated Asn is converted to Asp with mass shift of +1 Da, so such change serves as a clue of formerly glycosylated. However, deamidation (change from N to D) occurs frequently in vivo and in vitro, and deamidated sites are difficult to distinguish from trace marks of deglycosylation. Hence, we established a method for distinguishing glycosylated sites based on the difference in reaction rate between slow-progressing deamidation and fast-progressing enzymatic deglycosylation. When glycan removal was performed in solution composed of 18O-labeled water as the solvent, the heavy oxygen atom in the solvent was selectively incorporated to the glycosylation site, resulting in a heavier deglycopeptide (Figure 1). The authors first attempted to comprehensively register these glycosylation sites using this method.
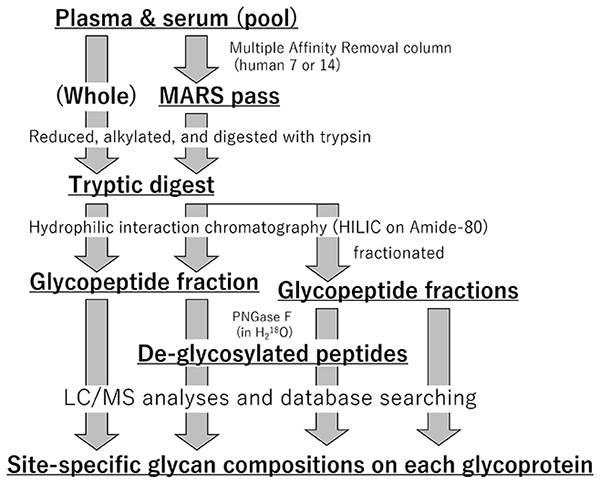
Albumin, a non-glycoprotein, and a few major (abundant) proteins in plasma/serum leads to difficulty with the identification of many other minor glycoproteins. We prepare both intact samples and depleted samples, and glycopeptides of both samples are enriched to identify glycosylation sites and glycan composition on each site (Figure 2). It is known that when obtaining an MS2 spectrum by data-dependent acquisition, repeated analysis increases the number of sites identified. In addition, flow rate, column choice, gradient time, and other factors may be changed to maximize efficiency and broaden the scale of measurement. In parallel, glycopeptide samples are fractionated to reduce complexity and analyzed in parallel for in-depth analysis. For example, if some major proteins composed of 90% of all serum proteins were first eliminated, the depth of analysis can be increased by one order magnitude compared with levels in intact serum. If the glycopeptide sample are fractionated to 10 different populations and analyzed the same way, the depth of analysis will be increased more one order magnitude. In the early stages of the project, we will work to obtain information on the glycosylation sites of 1,000 glycoproteins.
The difficulty with glycopeptide identification involves multiple stages. Reliable identification is hampered by low ionization efficiency, dispersion of molecular species, difficulty in fragmentation, and structural features that are prone to misinterpretation (assignment). The low ionization efficiency is due to the addition of large modifiers such as N-glycans and the attachment of one or more sialic acid units that add negative charges to the non-reducing ends of the glycans. One factor decreasing detection sensitivity is glycan heterogeneity. If we assume that 10 fmol of a protein is successfully digested by protease, then 10 fmol of each peptide product will be produced. If 10 different weight glycans attach to the peptide at the glycosylation site, the abundance of each glycopeptide will be 1 fmol on average. In the case of complex glycans, the addition of much more than 10 glycans unavoidably causes sensitivity reductions due to dispersion. Another problem is difficulty of fragmentation. MS-based identification depends on fragmentation. Peptides can be identified as a set of fragment ions resulting from peptide bond cleavage (b/y ions). A glycopeptide consists of an amino acid oligomer and monosaccharide oligomer(s) bound together, and glycosidic bonds in glycans are weaker than peptide bonds; therefore, weak (solitary) collision-induced dissociation breaks apart the glycan moiety but not the peptide, so that the peptide cannot be identified.
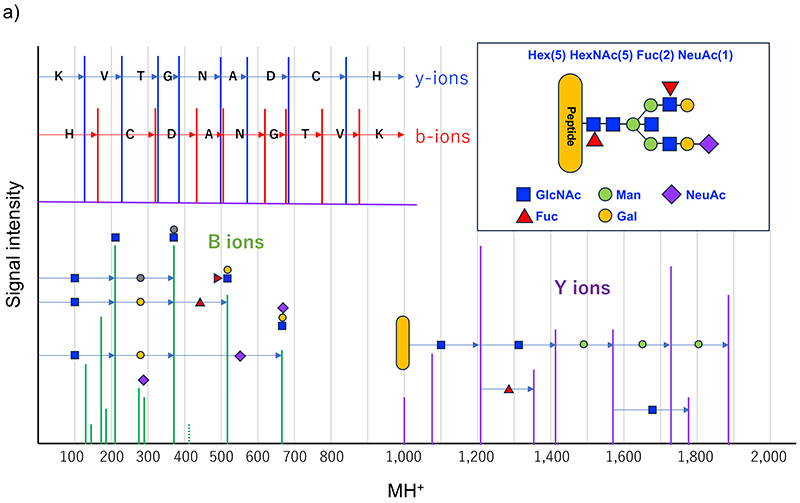
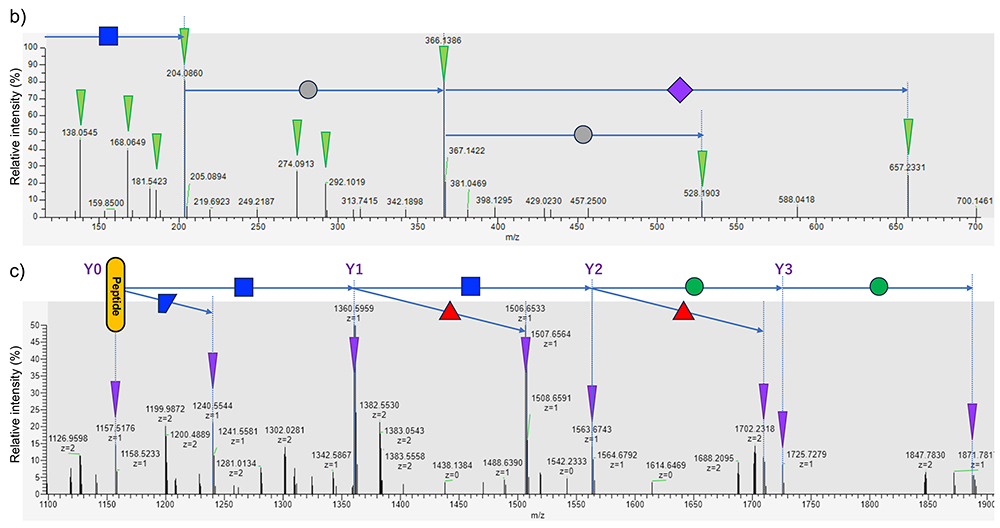
Recently, it has become possible to produce peptide fragment ions using higher energy collision-induced dissociation (HCD), which makes glycopeptide identification easier based on MS2. Because glycopeptides are diverse in sequence and glycan structure, they cannot be uniformly fragmented under constant conditions. Hence, various glycopeptides can be fragmented using the HCD method at more than one energy level of fragmentation and combining the resulting spectra to obtain an MS2 spectrum. An MS2 spectrum of a typical glycopeptide is shown in Figure 3. At low energy levels, the glycan glycoside bond is cleaved to produce ions of the peptide and saccharides close to the peptide (Y-series ions). In the case of N-glycans, a series of signals like peptide + GlcNAc + GlcNAc + Man + Man + Man is observed because of the presence of the trimannosyl core structure. Since the component cannot be determined to be GlcNAc or the like simply in terms of mass, it should be written HexNAc; however, taking the biosynthetic pathway into account, the above representation may be acceptable. In this series, the presence of peptide + HexNAc + dHex and peptide + HexNAc(3) + Hex(1) ions is useful, these signals suggesting the presence of core fucose and bisecting GlcNAc, respectively. Of the Y-series ions, the predominant one is the Y1 ion, which comprises one GlcNAc unit remaining attached to the peptide. If the Y0-Y1-Y2 ion is observed as a HexNAc mass difference, the mass of Y0, i.e., the peptide, is known; therefore, the MS/MS ion search method in proteomics is applicable to identify the peptide based on the b/y ion resulting from peptide fragmentation and this mass of Y0. Glycan fragment ions resulting from the cleavage of one or some glycoside bonds suggest the presence and structural characteristics of glycans and are also called diagnostic ions. The presence of HexNAc is strongly suggested by signals such as m/z 138, 144, 168, 186, and 204. Structures like Gal-GlcNAc- are suggested by a signal of m/z 366, though the signal is weak because Hex is unlikely to ionize. Additionally, ions derived from NeuAc (m/z 292 and 274), and NeuGc-derived ions (m/z 308 and 290) are helpful in distinguishing sialic acid. Besides monosaccharides, common NeuAc-Hex-HexNAc-(m/z657), as well as Hex(dHex)-HexNAc (Lewisx/y; m/z512) and HexNAc(2) (LacdiNAc; m/z407) suggest the structural motif contained and serve as sources of information for structural analysis. However, much attention should be paid to the likelihood that these diagnostic ions may have been derived from contaminating precursor ions. Intensified collision energy cuts peptide bonds to produce b/y ions, which facilitate peptide moiety identification. Many MS2 analysis software programs operate on an algorithm used to identify peptide sequences and glycan compositions (and small motif information) based on the above characteristics; however, the number of resulting fragments is not always sufficient for the identification, so that it is difficult to determine the accuracy of glycopeptide identification. Furthermore, some intrinsic structural characteristics cause glycan composition misidentification. The most difficult issue is the location of oxygen atoms. The elemental compositions of Hex and Fuc differ from each other by one oxygen atom, and the same applies to the difference between NeuAc and NeuGc. Therefore, since Hex + NeuAc = Fuc + NeuGc, multiple glycan compositions match one mass, making it difficult to identify which one is correct. Identification and confirmation may be possible by checking fragment ions, or by assuming the absence of NeuGc in the human sequence; however, identification algorithm failures are possible as evidence is not always available. Additionally, an erroneous determination of the monoisotopic mass of the precursor ion leads to misidentification due to the relationship Fuc(2)=NeuAc+1. In glycopeptides, particularly those consisting of a long peptide and a large glycan attached thereto, monoisotopic signal identification errors are not rare because the monoisotopic signal is very weak. Glycopeptides with multiple fucoses, specifically five Fuc molecules with a mass similar to that of 2*[Hex(1)HexNAc(1)], may be indistinguishable when they have large masses. As described above, there is still large room for developing software for identifying glycopeptides from MS2 spectra. In the Human Glycoproteomics Initiative (HGI) of the Human Proteome Organization (HUPO), a pilot study was started and is currently ongoing in its second stage6. Further development is expected.
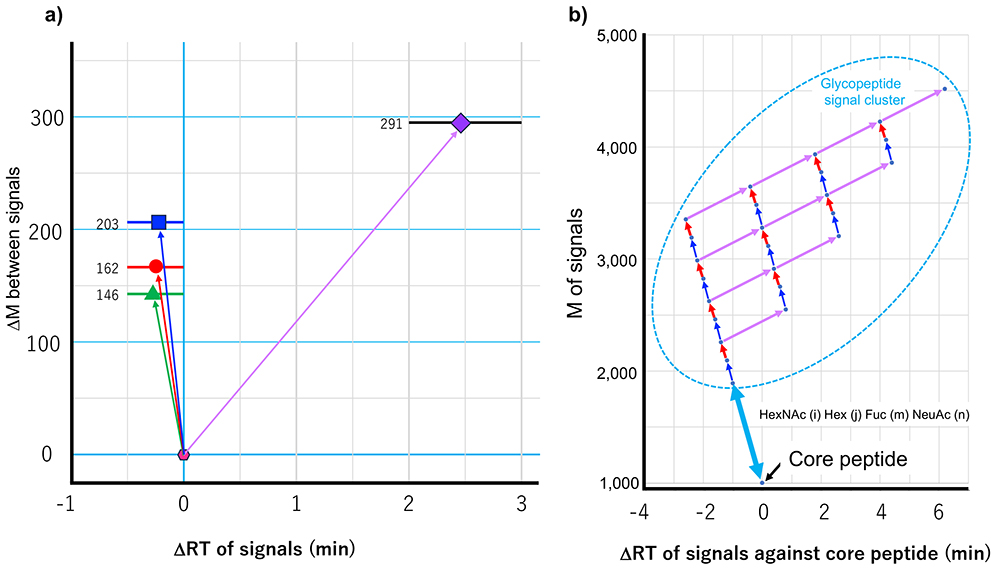
Now, more than 10,000 different proteins can be identified in samples containing extremely small amounts of protease digests of cell extracts (100-200 ng). In the case of glycoproteins, however, it is difficult to identify more than 2,000 proteins, even their glycosylation sites. This applies even more to glycan addition. Because the present project aims to increase the scale of glycopeptides identification beyond this barrier, higher sensitivity and quicker processing are essential. To this end, we are working to develop and apply a highly sensitive method of identification that does not depend on MS2, which is developed by the present authors, and to establish an identification technology based on the thus-obtained information (a method of reference-aided identification). The non-MS2-dependent method is a glycan heterogeneity-based, and glycopeptide elution profile-based method, which is named "Glycan heterogeneity-based Relational identification of glycopeptide signals on the elution profile", known as Glyco-RIDGE)7,8 (Figure 4). As described above, glycan heterogeneity reduces sensitivity in glycopeptide identification, and the Glyco-RIDGE method makes the best use of this feature. Specifically, the reverse-phase LC used in LC/MS separates glycopeptides mainly on the basis of the hydrophobicity of the peptide moiety; therefore, a set of glycopeptides with the same peptide moiety and different glycan compositions have similar retention times. In this process, retention time is slightly shortened by addition of a neutral saccharide (Hex, HexNAc, or Fuc) decreased by increasing the size of the glycan and increased by addition of an acidic saccharide (NeuAc). Furthermore, the mass differences between these glycopeptide signals appear to apply exclusively to one of four different monosaccharides (four types in terms of mass). Thus, for example, glycopeptides with Hex(5)HexNAc(2) and Hex(6)HexNAc(2) can be found as a pair of identical core glycopeptide signals. If one of the pair of signals is identified using MS2, the other is likely to be the same core glycopeptide. If a pair of signal appears to be a coincidence, a minimum number of clusters of three or four helps decrease coincidences. If none is identified in the detected cluster of signals, the signals can be assigned by comparing their masses with the mass of the deglycosylated peptide. Since the presence of non-glycosylated peptides (mass) and retention times are likely detected at high sensitivity using the above-described IGOT site mapping method, these parameters are able to utilize. It appears that some researchers are reluctant to identify glycopeptides on the basis of a combination of mass differences and retention time, without MS2 information. Of course, the provider does not want to spread incorrect information (identification results). In quantitative analysis in proteomics, especially when the MS2 spectra are data-dependently acquired, if a signal is identified in one sample and the corresponding signal is not identified in another sample, the identity of the signals is correlated based on retention time and mass, namely match-between-runs algorithm appears to be acceptable9. Although it may be not agreeable to handle the mass and mass differences in the same way, the present author considers it meaningful to provide possible data, while presenting reliable data (available supportive evidence). The author hopes that our approach will be accepted as a tool for finding clues to hidden truths.
In proteomics, data independent MS2 acquisition (DIA) has led to the explosive increase in the number of proteins identified. Initially, spectra library of previously matched was required for the identification; at present, however, peptide elution positions can be predicted, and proteins can be identified based on the agreement between the calculated precursor mass and the calculated fragment ion masses. Likewise, it is possible to predict the elution position of glycopeptides. However, as described above, glycopeptides even with different glycans are eluted in close retention time, and the HCD-produced fragments are nearly the same, therefore, it is difficult to link the precursor and fragment ions based on the elution profile (mass chromatogram), and it may be difficult to increase identification sensitivity due to MS2 dependence, even if the identification is not data-dependent. However, since MS equipment is becoming increasingly more sophisticated, capable of more sensitive and rapid measurement, we may be optimistic that glycans will be easily identifiable in the near future.
On the other hand, tissue-specific analysis yields only obscure data because glycans are variable depending on the type of cell. Ultimately, single-cell observations may be required. The present study is being conducted with the aim of establishing an in-depth information system that can be referenced in advance for use in future analyses. The results of the Genome Project have made it unnecessary to sequence and identify peptides using Edman degradation, to clone the cDNA of the protein of interest over many years, and to determine base sequences using the Maxam-Gilbert method or the Sanger method. With this successful story in mind, the present author would like to establish a platform for easily identifying glycopeptides. Of course, human glycans include many kinds and structures that are not detectable using the present approach. For example, oligo- and poly-sialic acids (sialic acid units joined together) are difficult to detect and may collapse under the sample preparation conditions we use; therefore, analyses are ongoing using other approaches. Furthermore, as described at the beginning of this paper, the present approach only allows the determination of glycan compositions; however, we are planning to acquire site-specific information on the choices, binding positions, and orientations of monosaccharides. Finally, while informaticians may be reluctant to develop new analytical tools as they lack research novelty, we want the cooperation of information scientists who will work with us on this task.



