
Go Hirai
Professor, Graduate School of Pharmaceutical Sciences, Kyushu University
After graduating from the Department of Chemistry, Faculty of Science, Tohoku University in 1997, Go Hirai completed his doctoral studies in Chemistry at the same university in 2002, earning a Ph.D. under the supervision of Prof. Masahiro Hirama. After working as an assistant professor in the Institute of Multidisciplinary Research for Advanced Materials at Tohoku University (with Prof. Mikiko Sodeoka), he became a research scientist at the Synthetic Organic Chemistry Laboratory, RIKEN led by Prof. Sodeoka in 2004. He was promoted to Senior Research Scientist in 2010. He has been in his current position since 2016.
His achievements include the Incentive Award from the Society of Synthetic Organic Chemistry, Japan (2013), and the GlycoTOKYO Award (2014).
Replacing the native O-glycosidic linkage in glycans with a C-glycosidic linkage can create analogs resistant to glycoside hydrolases without significantly altering the overall structure. The C-glycosidic linkage also allows the introduction of substituents, enabling the construction of diverse glycan architectures. This paper introduces glycan analogs linked via a simple CH2 group as well as those linked through a CHF group. These three types of analogs (including diastereomers) possess similar steric characteristics but distinct electronic properties. We summarize current knowledge on synthetic methodologies for "linkage-editing" and discuss its impact on the biological functions of glycans.
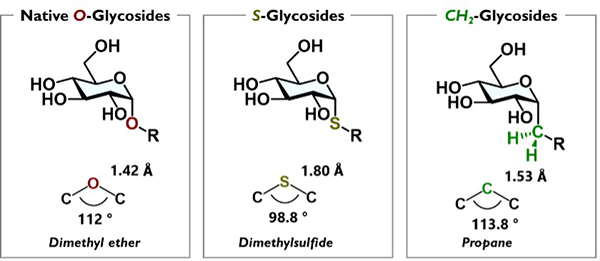
Glycans and glycoconjugates contain monosaccharide or aglycone units connected by O-glycosidic linkages1. Glycoside hydrolases cleave these O-glycosidic linkages in accordance with their substrate specificity, leading to glycan degradation and functional changes that are essential for cellular homeostasis. In drug discovery applications, this degradation equates to a loss of biological activity, prompting the development of glycan analogs resistant to glycoside hydrolases. Early analogs were created by replacing O-glycosidic linkages with S-glycosidic linkages2, which, while efficient from a synthetic perspective, affords structures with notably different bond lengths and angles, raising concerns about their validity as true analogs. From the viewpoint of structural similarity, the C-glycosidic linkage is the most rational choice. Our laboratory refers to these C-glycosidic linkage-containing analogs as C-glycoside (carbon-linked) pseudo-glycans.
In 2004, Franck and co-workers synthesized a CH2-linked α-galactosylceramide analog (pseudo-α-GalCer, CH2-α-GalCer, Figure 2), which exhibited anti-tumor activity approximately 100 times higher and anti-malarial activity roughly 1,000 times higher than the corresponding O-linked α-GalCer in vivo3,4. This clearly demonstrated the potential of C-glycoside pseudo-glycans as drug leads. However, synthesizing C-glycoside pseudo-glycans typically involves numerous synthetic steps. Although simple monosaccharide analogs with a C-glycoside linkage or pseudo-glycans with a C-glycoside linkage at the reducing end, which is not cleaved by exo-glycoside hydrolases, have been reported, the development of glycoside hydrolase-resistant pseudo-glycans with C-glycosidic linkage replacing the cleavable O-glycoside linkage still remains at an early stage5–7. Furthermore, functional studies have primarily been limited to analyzing interactions with lectins, and there has been little study of the effects on biological activity or function, except for CH2-α-GalCer8,9.
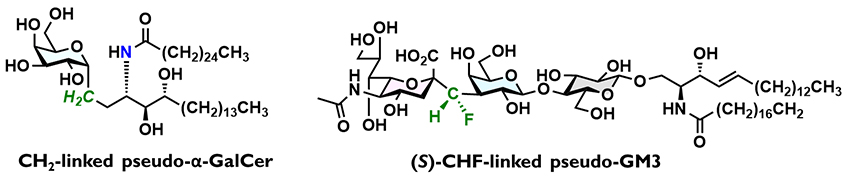
Our initial efforts focused on creating C-glycoside pseudo-glycans based on ganglioside GM310–12. Since the degradation of GM3 starts with cleavage of the glycosidic linkage of sialic acid by sialidase, we replaced this linkage with a C-glycoside (Figure 2). We considered not only a conventional CH2-linkage but also a fluorine-containing CF2- or CHF-linkage, since fluorine atom(s) would modulate the acidity of sialic acid and influence the conformation of the glycosidic linkage. Evaluation of the biological activity of four pseudo-GM3 analogs revealed that the CHF-linked analog exhibited higher biological activity than native GM3, with the (S)-CHF linkage displaying slightly higher activity than the (R)-CHF linkage. The CF2-linked compound has an acidity similar to that of native sialic acid, and the acidity decreased in the order of CF2-linked, CHF-linked, and CH2-linked types. On the other hand, the CH2-analog, with no stereoelectronic effect, has a higher flexibility in terms of rotation of the glycosidic linkage, while introducing an F atom restricts the conformation due to the gauche effect13 and dipole repulsion. We considered that the (S)-CHF-linked analog achieves high biological activity due to its appropriate acidity and stabilization of an active conformation. This is in line with the widely accepted concept in medicinal chemistry that glycans exert their biological activity by interacting with other biomolecules in a specific conformation.
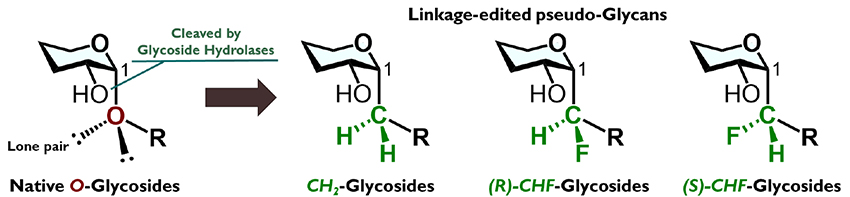
We use the term linkage-editing strategy to describe the design of pseudo-glycans by varying the glycosidic linkage (Figure 3). We anticipated that application of this strategy to various glycans would afford glycoside hydrolase-resistant pseudo-glycans that exhibit high biological activity through conformational regulation.
Is the premise that a specific conformation contributes to the biological activity of native glycans correct?
The O-glycosidic bond is a single bond, and its rotation affords considerable molecular flexibility14. Nevertheless, steric/electrostatic repulsion and the stereoelectronic effect of the O-glycosidic bond (the exo-anomeric effect) are expected to favor particular conformation(s)15, and it is plausible that a thermodynamically more stable conformation(s) is involved in interactions with biomolecules to elicit biological activity. However, due to the conformational flexibility, interactions in a specific conformation are entropically unfavorable, meaning that biological systems bear the cost of manifesting biological functions. Thus, there may be a particular reason why nature selected conformationally flexible glycans as the third chain of life.
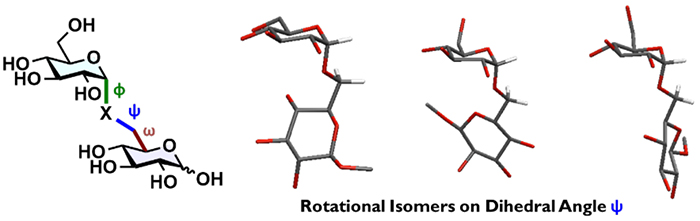
If conformational flexibility and conformational distribution (Figure 4), rather than a stable conformation(s), are critical for biological activity, the potential significance of linkage-editing is dramatically altered. For example, CH2-linked pseudo-glycans should have a broader conformational distribution than the corresponding native glycans, even though they are structurally similar, due to the absence of stereoelectronic effects. In contrast, CHF-linked pseudo-glycans, constrained by the fluorine atom, would show a biased conformational distribution, depending on their stereochemistry. The different conformational distributions of native glycan and C-glycoside pseudo-glycan should lead to changes in molecular dynamics, ultimately resulting in altered biological activity. Enhanced activity as in pseudo-GM3, activity loss, and even new phenotypes are all possibilities. In other words, we have come to view the construction of C-glycoside pseudo-glycans as a strategy for creating novel bioactive molecules rather than merely for obtaining glycan mimetics resistant to degradation.
We aimed to synthesize linkage-edited pseudo-glycans corresponding to various glycan structures to test the above ideas. Developing efficient synthetic methodology is critical for this purpose. We focused on establishing a direct C-glycosylation approach that enables the formation of C-glycosidic linkage via intermolecular coupling reactions while retaining functional groups in glycans and glycoconjugates. Our efforts since 2016 have been aimed at stereoselective pseudo-glycan synthesis through direct C-glycosylation utilizing anomeric radicals16–18.
Here, we describe an example of a reductive coupling reaction using glycosyl bromide as the donor and an acceptor incorporating a bromofluoroolefin group. We optimized the reaction conditions based on the reductive coupling reaction reported by MacMillan and co-workers, which utilizes Ir photocatalyst, Ni catalyst, and (TMS)3SiOH as a halogen atom transfer (XAT) reagent19,20. When using donor 1 with cyclic carbonate protection, α-selective coupling reaction with acceptor 2 proceeded to provide the α-fluorovinyl-C-glycoside 3 in high yield (Figure 5). Carbonate protection fixes the conformation of the resulting anomeric radical species in a chair form. At this point, the anomeric effect favors the formation of an α-oriented (sp3) radical 4, which is expected to result in high α-selectivity21–23. In contrast, irradiation of donor 5 and acceptor 2 under similar conditions afforded product 6 with significantly reduced product yield and stereoselectivity. In the case of donor 5, the obtained radical species 8 adopts a boat conformation, directing the spin in both α and β orientations (sp2 radical) via a quasi-homo-anomeric effect. Additionally, it is known that the 2-position hydroxyl group, protected by an acyl group, undergoes acyloxy rearrangement, leading to the formation of the thermodynamically stable secondary radical 9. In fact, the formation of product 7, derived from radical 9, indicates that cyclic carbonate protection plays an essential role in determining both the reactivity and stereoselectivity.
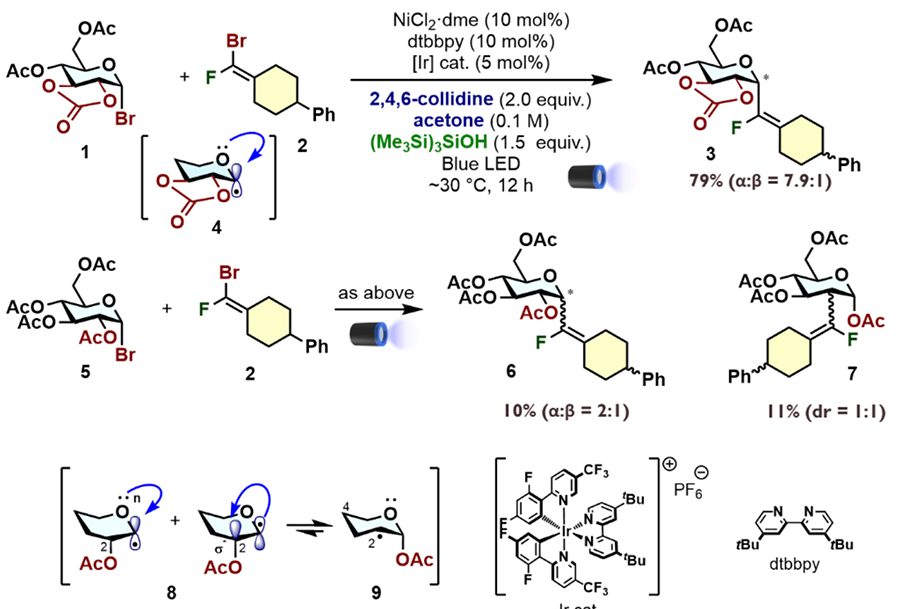
This method enables the synthesis of various α-fluorovinyl C-glycoside pseudo-glycans. While only the 1,6-linked disaccharide analog is shown in Figure 6 as an example, the approach can be applied to various linkage types. Notably, when using a galactose donor, cyclic protective groups play a significant role, whereas high α-selectivity is achieved even without cyclic protection when using mannose donors.
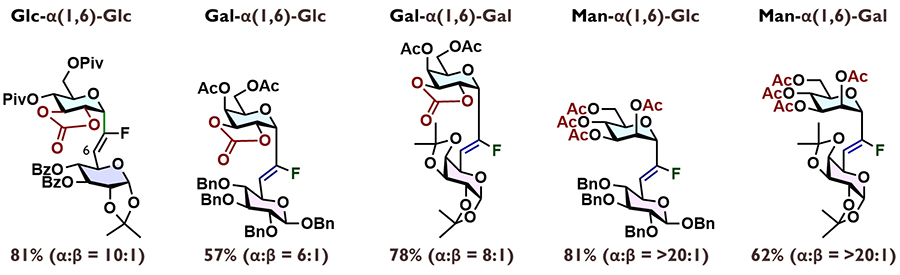
Through hydrogenation of the fluoroolefin and subsequent deprotection, C-glycoside pseudo-glycans can be synthesized. Here, we describe the synthesis of C-glycoside pseudo-α-GalCer in detail.
The coupling reaction between galactose donor 10 and sphingosine-derived acceptor 11 affords compound 12 with complete α-selectivity. The cyclic carbonate is removed to obtain diol 13. Hydrogenation in the presence of triethylamine and a Pt/C catalyst results in the reduction of both the double bond and the C-F bond, enabling the construction of the CH2-linkage. Removal of the Boc group, introduction of an acyl group on the resulting amine, and subsequent deprotection afford the CH₂-linked pseudo-α-GalCer (CH₂-α-GalCer). When the catalyst in the initial hydrogenation was changed to Pearlman's catalyst (Pd(OH)2/C), the (S)-CHF linkage was obtained with high stereoselectivity (7.6:1). This intermediate was converted to (S)-CHF-α-GalCer in three steps. In contrast, the selective synthesis of the (R)-CHF linkage proved challenging, but we eventually found that hydrogenation of 13 using Crabtree's catalyst preferentially affords the (S)-CHF-linked analog with a selectivity of 3.3:1. Using the minor product obtained, we achieved the synthesis of (R)-CHF-α-GalCer through a similar three-step process. Although further improvement is still needed, each linkage was constructed by means of late-stage diversification, allowing us to complete the synthesis of three types of pseudo-α-GalCer.
α-GalCer is presented by CD1d receptors on various antigen-presenting cells. Recognition of this complex by the T-cell receptor (TCR) on NKT cells activates these cells to produce various cytokines. The effect varies depending on the balance of cytokines produced; for example, the production of Th1 cytokines such as IFN-γ promotes cellular immunity, leading to antitumor effects24. Consequently, α-GalCer is considered as a lead compound for potential therapies targeting various diseases, and numerous derivatives have been investigated25. Among them, CH2-α-GalCer showed a potent antitumor effect in vivo, suggesting that introducing degradation resistance leads to a significant enhancement of activity in vivo. Conversely, it was reported that the switch from an O-glycosidic to a CH2-glycosidic linkage reduced the stability of the ternary complex with CD1d-TCR26, and that mechanisms independent of NKT cell activation exist27. In addition, Mootoo and colleagues synthesized (R)-CHF-α-GalCer and reported its function in collaboration with Ma and co-authors28. Though (R)-CHF-α-GalCer is presented by CD1d, like α-GalCer, it completely blocks the production of IFN-γ and IL-4 when co-administered with CH2-α-GalCer in mice, though these cytokines are induced by CH2-α-GalCer alone. (R)-CHF-α-GalCer also selectively induces the production of IL-17 when administered alone.
In this study, by successfully synthesizing three types of C-glycoside pseudo-α-GalCer, we were able to conduct a comparative evaluation of the linkage-editing effects within the same assessment system. We focused on the effects on immunostimulatory activity and NKT cell activation.
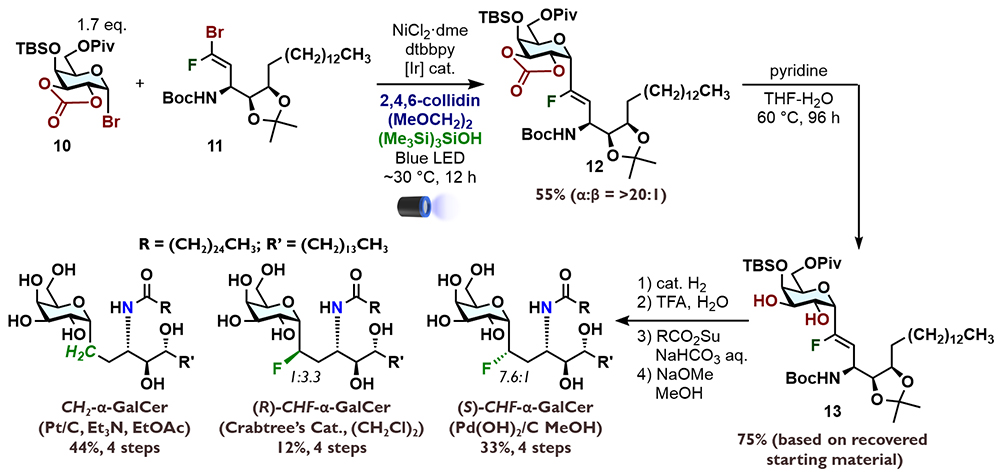
Given the immunostimulatory activity of α-GalCer, its adjuvant activity has also been utilized to enhance vaccine efficacy. With the support of Prof. Jun Kunisawa (National Institutes of Biomedical Innovation, Health and Nutrition), we evaluated the adjuvant activity of pseudo-α-GalCer. Mice were administered OVA as the antigen with either 2 μg of α-GalCer or pseudo-α-GalCer, and anti-OVA antibodies (IgG and IgM) in serum were measured two weeks later by ELISA. While sufficient antibody production was observed in α-GalCer-treated mice, CH2-α-GalCer generated only about half the native level, while both CHF-α-GalCer isomers induced minimal antibody production (Figure 8A). Initially, we anticipated that one of the stereoisomers of CHF-α-GalCer might be stabilized in an active conformation due to conformational control by the fluorine atom, so the low activity was disappointing. We next quantified cytokines produced by splenocytes treated with C-glycoside pseudo-α-GalCer using a cytometric bead array kit. CH2-α-GalCer induced production of IFN-γ, IL-4, and IL-17A, though at lower levels than were obtained with α-GalCer, while CHF-α-GalCer showed negligible cytokine production. Although the molecular structures are similar, it appears that linkage-editing of α-GalCer in this way led to a significant decrease in immunostimulatory activity.
As was the case for the reported ternary complex of CH2-α-GalCer with CD1d-TCR26, converting the linkage to a CHF group did not appear to prevent the formation of the ternary complex. Ma and colleagues demonstrated that (R)-CHF-α-GalCer is presented by CD1d; however, the effect on NKT cell activation was not established. Therefore, with the collaboration of Prof. Sho Yamasaki (Osaka University), we investigated whether C-glycoside pseudo-α-GalCer activates NKT cells. We found that none of the C-glycoside pseudo-α-GalCer derivatives activated NKT cells. This result might be related to the fact that CH2-α-GalCer induces sufficient cytokine production in vivo but induces little in vitro. Furthermore, the pronounced antitumor activity of CH2-α-GalCer and the IL-17 induction by (R)-CHF-α-GalCer are characteristic functions observed in vivo, and may be independent of NKT cell activation.
Thus, we considered the possibility that C-glycoside pseudo-α-GalCer might inhibit NKT cell activation. Indeed, we found that C-glycoside pseudo-α-GalCer inhibited NKT cell activation induced by α-GalCer (Figure 8B). The effect of (R)-CHF-α-GalCer was particularly potent, indicating that linkage-editing had converted α-GalCer's biological activity from agonistic to antagonistic. It remains unclear whether the CHF-linked analog can form a ternary complex with CD1d and TCR, but even if it binds similarly to α-GalCer or CH2-α-GalCer, differences in hydrophobicity and conformational distribution are expected to alter the dynamics of the ternary complex. This is consistent with the idea that the conformational distribution of α-GalCer plays an important role in NKT cell activation, i.e., that changes in the populations of glycolipid conformers can dramatically influence the activity29.
In addition to pseudo-α-GalCer analogs, other C-glycoside pseudo-glycans also show unique changes of activity. For example, CH₂-linked analogs of pseudo-isomaltose exhibit enhanced activity, whereas both stereoisomers of CHF-analogs lack activity29. In the case of pseudo-melibiosamine, CH2-analogs showed slightly reduced activity, while (S)-CHF-analogs almost lacked activity, and (R)-CHF analogs showed approximately five-fold increased activity30. We have also synthesized two other pseudo-glycoconjugates, whose biological activity also varies depending on the linkage. We attribute these activity changes to differences in glycan conformational distribution and glycan dynamics, but this hypothesis remains to be confirmed. Nevertheless, whatever the mechanism, the differing biological activities despite the overall structural similarity, combined with degradation resistance, mean that these compounds offer significant advantages as molecular tools in chemical biology research. Further work along this line is in progress.



