
Masaki Kurogochi
Research Scientist
Institute for Glyco-core Research, Nagoya University, Tokai National Higher Education and Research System
Masaki Kurogochi graduated from Hokkaido University in 1998, got a doctoral degree in science. He worked as a senior researcher in Northern Advancement Center for Science and Technology(2004), a postdoctoral researcher in Hokkaido University (2005), became an assistant professor at the Faculty of Advanced Life Science, Hokkaido University. In 2012, he joined The Noguchi Institute, where he worked as a research scientist in Laboratory of Glycobiology, a leader in Laboratory of Glyco-bioengineering, a research scientist in Laboratory of Glyco-organic Chemistry. He has been in his current position since 2024. His research interests are focused on organic chemistry, analytical chemistry, glycoengineering, glycomics and glycoproteomics.

Jun-ichi Furukawa
Designated Professor
Institute for Glyco-core Research, Nagoya University, Tokai National Higher Education and Research System
Visiting Professor
Department of Orthopaedic Surgery, Faculty of Medicine and Graduate School of Medicine, Hokkaido University Jun-ichi Furukawa graduated from Faculty of Science at Hokkaido University in 1996, received his Ph.D. in Environmental Earth Science at Hokkaido University in 2001. Then, he worked as a JSPS postdoctoral, a postdoctoral fellow (2002–2006), and as a research assistant professor (2006–2016) in Faculty of Science, Hokkaido University. In 2016, he was a Specially Appointed Associate Professor of Faculty of Medicine, Hokkaido University. He has been in his current position since 2022. His current research interest include bioorganic chemistry and analytical chemistry focused on glycans. He is studying the comprehensive glycomic analysis using his original analytical tools.
In segment 2 of the Human Glycome Atlas Project (HGA), the generation of a large-scale human glycome catalog (total human plasma glycome) is one of the most important missions for constructing a database called TOHSA. In this section, we introduce our latest studies, including sample preparation for the analysis of O-glycans in human plasma/serum.
More than 50% of proteins in living organisms are glycoproteins attached to glycans. Glycans on the glycoproteins affect protein folding and structural stability, and they play important roles in the transport, secretion, recognition, metabolism, and signal transduction of biomolecules. These glycans are widely classified into N-glycans and O-glycans. N-Glycans are attached to the side chain of asparagine, and O-glycans are attached to the side chain of serine or threonine. Previously, O-glycans were thought to be a rare post-translational modification present in mucins and mucin-like proteins in eukaryotes, but in recent years they have been found to be an abundant post-translational modification present in approximately 80% of secreted proteins1,2. As structural changes in O-glycans have been observed in diseases such as IgA nephropathy3, Tn syndrome4, Crohn’s disease5, and tumorigenesis6, much research has been performed to elucidate the biological and clinical functions of O-glycans. Although analysis of N-glycans, for which there are enzymes that be used to comprehensively cleave the N-glycans from proteins, has been progressing, it is necessary to cleave O-glycans by chemical methods because there are no enzymes that comprehensively cleave them from proteins. We have reported a method for simultaneously cleaving O-glycans from glycoproteins by β-elimination and labeling the released O-glycans by introducing a pyrazolone reagent into the aldehyde group at the reducing end. (The alkaline β-elimination and pyrazolone labeling method are called the BEP method)7. At present, we are developing an improved technique by combining the evaporative BEP method with the sialic acid linkage-specific alkylamidation method (SALSA), which can discriminate sialylated glycan isomers by using mass spectrometry, for the analysis of O-glycans in human plasma/serum8.
In segment 2 of the Human Glycome Atlas Project (HGA), the generation of a large-scale human glycome catalog (total human plasma glycome) is one of the most important missions for constructing a database called TOHSA. In this section, we introduce our latest studies, which consist of 2-1) enhancement of the O-glycan yield by using an evaporative BEP method; 2-2) the highly sensitive detection of sialyl O-glycans in MALDI-TOF MS measurements by using the SALSA method; and 3) sample preparation and analysis of O-glycans in human plasma/serum for large-scale study.
O-Glycosylation is one of the most common post-translational modifications of proteins, and O-glycans are attached to the side chain of a Ser or Thr residue. O-GalNAc glycans (called mucin-type O-glycans) are widely classified into eight types, from core type 1 to core type 8, on the basis of their characteristic structures. All types of N-glycans (high-mannose type, hybrid type, complex type) can be comprehensively cleaved from proteins by using commercially available peptide-N-glycosidase F (PNGase F), whereas for O-glycans, although there are endoglycosidases that can cleave the core 1 and core 3 types, there is no enzyme that can comprehensively cleave all types of O-glycans. Therefore, for comprehensive analysis of the O-glycans, we need to cleave them by using chemical methods. Under alkaline conditions, release of an α-proton from a Ser or Thr residue leads to the cleavage of O-glycans attached to the side chains (β-carbon position) of Ser and Thr residues (alkaline β-elimination). This β-elimination reaction successfully releases O-glycans comprehensively, but it also causes a peeling reaction that generates degradation of the O-glycans. Therefore, a method of cleaving O-glycans that overcomes the peeling reaction is required. We have developed a BEP reaction7,9,10 that can suppress the peeling reaction by labeling the aldehyde group at the reducing end of the released O-glycans with a pyrazolone compound at the same time as the O-glycans are cleaved under alkaline conditions. This BEP method was initially performed at 75 ºC for 16 h in a closed system, but the O-glycan yield was then improved by using a microwave reactor at 110 ºC for 2 h in a closed system11. At present, considering the versatility and simplicity of the reaction device, we have developed an evaporative BEP method performed at 105 ºC for 2 h in an open system, and we have succeeded in further improving the O-glycan yield8.
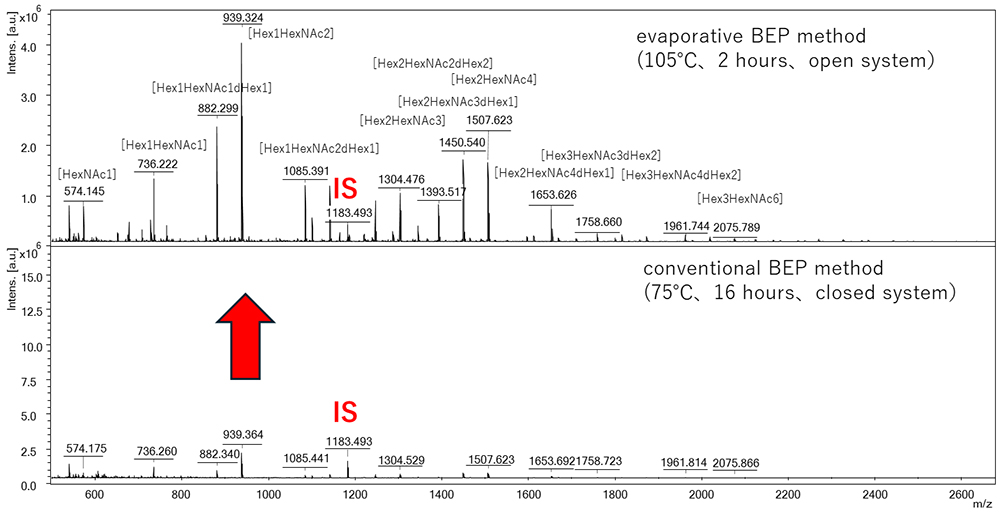
Here, by using the same sample (porcine stomach mucin, PSM), a conventional BEP reaction (75 ºC, 16 h, closed system) and an evaporative BEP reaction (105 ºC, 2 h, open system) were performed, and the O-glycans prepared were measured by MALDI-TOF MS after the addition of an internal standard. Comparison of the main O-glycan peaks in the MS spectra revealed that the O-glycan yield was dramatically improved (Figure 1). This is likely because of acceleration of the reaction as a result of the high temperature, along with an increase in the reactant concentration associated with evaporation of the solvent.
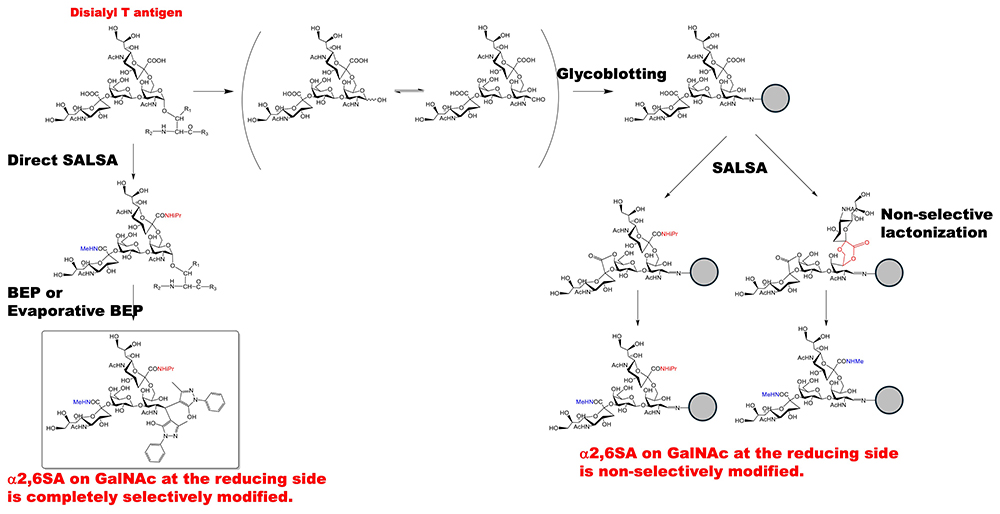
Recent studies have reported several unique sialic acid linkage-specific derivatizations for distinguishing glycan isomers by MS analysis12. We have developed a method of sialic acid linkage-specific derivatization by amidation with molecules that have different molecular weights13,14. This method is applicable to the released N-glycans and released glycosphingolipid glycans. They are captured on hydrazide beads by utilizing the fact that free glycans have an aldehyde group at the reducing end; this is called the glycoblotting method. The carboxylic acids of α2,6-linked sialic acid on the glycans is converted to isopropylamides, whereas those of the α2,3- and α2,8-linked sialic acids on the glycans form intramolecular lactones. The α2,3- and α2,8-linked sialic acids on the glycans are then converted to methylamides via the intramolecular lactones. This is called the SALSA method. When O-glycans were captured on a solid phase and subjected to the SALSA reaction in the same manner as described above, we found that the α2,6-linked sialic acid bound to the GalNAc residue on the reducing end of the disialyl T antigen in the O-glycan was converted to a mixture of isopropylamidated and methylamidated15. This is because the GalNAc at the reducing end captured on the solid phase is opened, and the OH group at the 5-position of GalNAc and the carboxy group of the sialic acid at the α2,6-linkage are nonselectively lactonized, followed by methylamidation (Figure 2, right). Therefore, to achieve selective amidation of the disialyl T antigen, it is necessary to perform the SALSA reaction on a protein in which the reducing-terminal GalNAc exists in a closed ring state, and to follow this with the evaporative BEP reaction (Direct SALSA, Figure 2, left)8. As described above, we performed SALSA to prepare methylamidated α2,3-linked monosialyl T antigen and isopropylamidated α2,6-linked monosialyl T antigen.
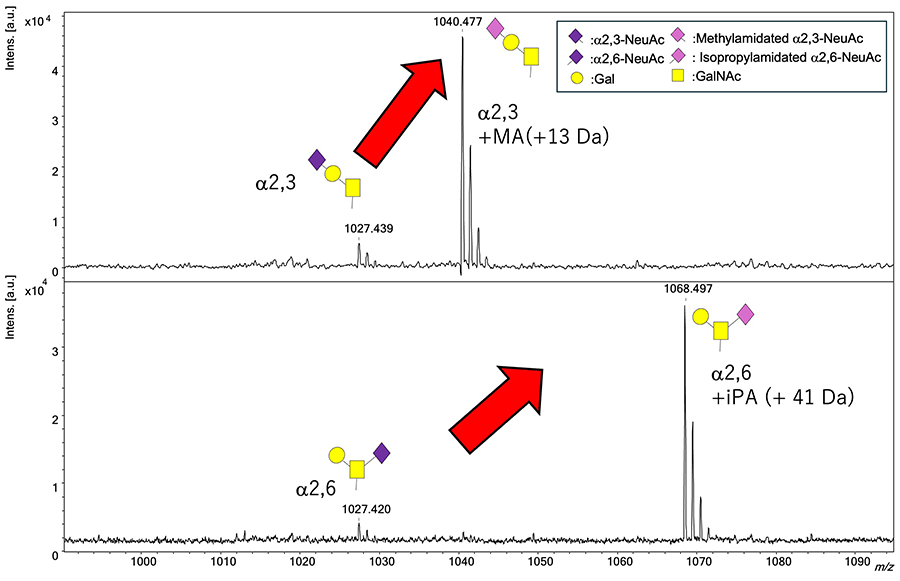
To show the advantages of the SALSA method, we performed MALDI-TOF MS measurements on a mixture of equal amounts of unmodified α2,3-linked monosialyl T antigen and SALSA-treated α2,3-linked monosialyl T antigen, as well as on a mixture of equal amounts of unmodified α2,6-linked monosialyl T antigen and SALSA-treated α2,6-linked monosialyl T antigen (Figure 3). We found that both α2,3-linked monosialyl T antigen and α2,6-linked monosialyl T antigen modified by SALSA dramatically enhanced the intensities of the glycan peaks in the respective MALDI-TOF MS spectra. This makes it possible to detect, with high sensitivity, differences in the linkage positions of sialic acids in sialic acid-containing glycans as differences in molecular weight.
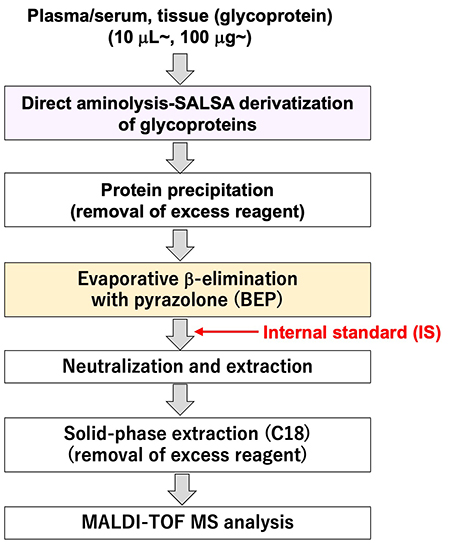
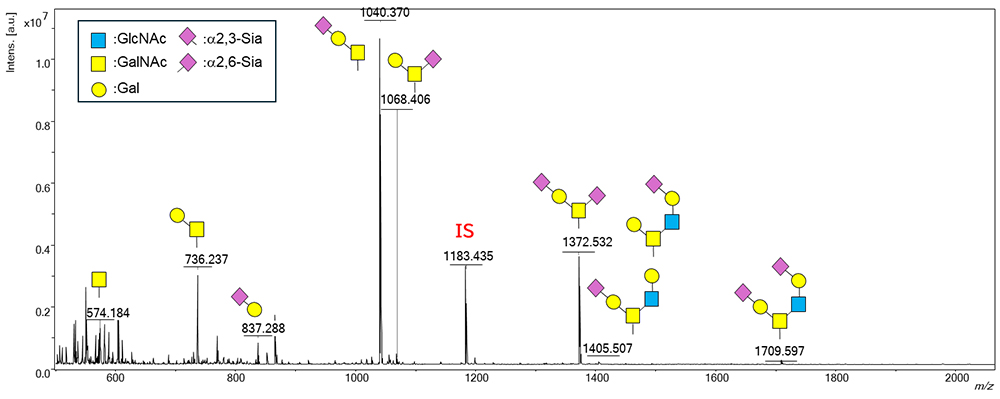
As described above, we are now ready to analyze O-glycans and have created a protocol for O-glycan analysis to perform a large-scale analysis in human serum/plasma. The protocol for the O-glycan analysis is shown in Figure 4. This protocol takes only a short time (about 4 h in total). Therefore, we believe that it has resolved the issues of sample preparation time and measurement time, which must be considered in large-scale analyses. Figure 5 shows the MS spectrum of O-glycans in commercially available serum. From this analysis, we could identify seven types of O-glycans containing different linkage positions of sialic acid in human serum.
We have finished developing a protocol for the analysis of O-glycans in serum/plasma in large-scale cohort studies, and we are currently improving the accuracy and sensitivity of this quantitative analysis of O-glycans. We are also developing an automated sample preparation system capable of processing multiple samples; this is essential for achieving comprehensive glycomics in large-scale cohort studies.



