
Ken Hanzawa
Research Scientist
Institute for Glyco-core Research, Gifu University, Tokai National Higher Education and Research System
Ken Hanzawa graduated from the Faculty of Science, Niigata University, in 2012 and received his Ph. D. degree in science from the Graduate School of Science and Technology, Niigata University, in 2017. Then, he worked as a Staff Researcher at the Research Center, Osaka International Cancer Institute (2017–2022). He has been in his current position since 2022. His current research interest focuses on high-throughput glycoproteomics and cancer biomarkers.

Kazuki Nakajima
Associate Professor
Institute for Glyco-core Research, Gifu University, Tokai National Higher Education and Research System
Kazuki Nakajima graduated from Kinki University in 2001 and received his Ph. D. degree in pharmacy, from the Graduate School of Pharmacy, Kindai University, in 2006. He worked as a Special Postdoctoral Researcher at RIKEN (2006–2009), as a Specially Appointed Assistant Professor and Researcher in the Systems Glycobiology Research Group, Osaka University and RIKEN (2009–2013), as a Researcher in the Brain Science Institute, RIKEN (2013–2016), and as a Senior Assistant Professor in the Support Center for Research Promotion, Fujita Health University (2016). He has been in his current position since 2022. His current research interest focuses on development of systems for high-throughput and automated glycoproteomics analysis. He is also interested in glycan biomarkers for kidney and neurodegenerative diseases.
The Human Glycome Atlas Project (HGA), which launched in April 2023, aims to perform glycoproteomics on a large cohort of samples, using a detailed human glycan map as a reference, to create a catalog of human glycans related to disease. During the first 5 years of the project, the focus will be on dementia and aging, and 20,000 samples mainly from plasma and serum will be analyzed. In the following 5 years, the scope will be broadened to include various diseases, and 200,000 samples will be targeted for analysis. Our goal is to establish and fully automate a global standard method for analyzing glycoproteomics profiles, enabling the large-scale analysis of glycan structures and the collection of comprehensive glycan data.
Technological advancements in artificial intelligence and robotics have led to a global trend towards the use of automation to expedite research processes. This trend was accelerated by the widespread adoption of automated polymerase chain reaction testing during the coronavirus disease 2019 pandemic, driven by the need to handle clinical samples safely and efficiently while minimizing infection risks. Automation offers several benefits, including increased sample processing capacity, reduced reliance on skilled technicians, and minimized risk of sample mis-selection.
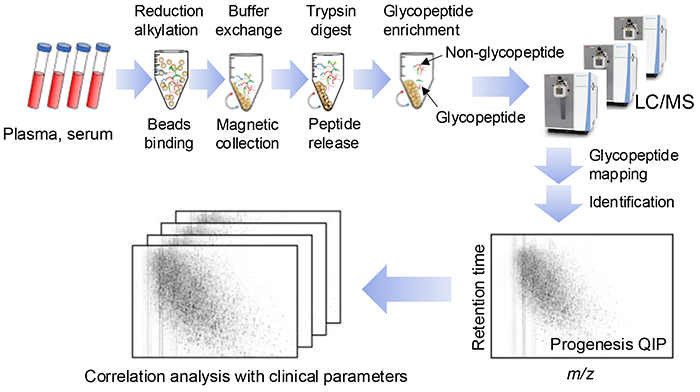
Proteomics and glycomics involve time-consuming, tedious, complex, multi-step processes. To automate these processes, robotic liquid handler platforms are employed1. However, current experimental procedures are still performed semi-automatically and intermittently. Applying automated protocols to blood samples poses many challenges to glycoproteomics analysis. The sample preparation process, similar to that with proteomics, involves protein extraction from blood samples, followed by reductive alkylation, peptide fragmentation, glycopeptide purification, drying, resolubilization, and dispensing the samples onto 96-well plates (Figure 1). A primary challenge in glycoproteomic multi-sample analysis is the extreme heterogeneity of glycopeptide samples. However, in multi-sample analysis, it is desirable to analyze each sample in a single run. Thus, we need to consider sample preparation methods that increase analytical depth and throughput.
Another challenge is the low ionization efficiency of glycopeptides in mass spectrometry (MS) measurements, resulting in low detection sensitivity. The presence of many residual non-glycosylated peptides also suppresses ionization of glycopeptides. Therefore, to comprehensively detect glycopeptides, non-glycosylated peptides must be removed as much as possible to prepare highly pure glycopeptide samples. Additionally, blood protein concentrations range from mg levels of albumin to trace amounts of cytokines, which requires the removal of albumin, immunoglobulin, and other abundant proteins in advance. Recently, a robust plasma glycoproteomic method has been established to overcome these challenges2,3.
Regarding the data analysis in our method, Progenesis QIP (Nonlinear) Note 1) was used to plot the data as a two-dimensional map showing the point with the m/z value and retention time of each glycopeptide, and the reference map-aided glycopeptide identification tool was used to identify the glycopeptide structure in the human glycan map (Figure 1). Additionally, Byonic Note 2) was used to plot tandem mass spectrometry (MS/MS) data for glycopeptide identification, and comparative quantification of the glycopeptides was based on peak intensity measurements. Our objective was to develop a comprehensive, high-throughput, and automation-friendly method for a large-scale cohort analysis. This article will detail our current efforts in N-glycoproteomic pretreatment methods as part of the HGA, along with automation challenges and equipment development.
Note 1) Progenesis-QIP is a liquid chromatography-mass spectrometry (LC-MS) data analysis software used for discovering significantly changing compounds.
Note 2) Byonic is a search engine for comprehensive peptide and protein identification.
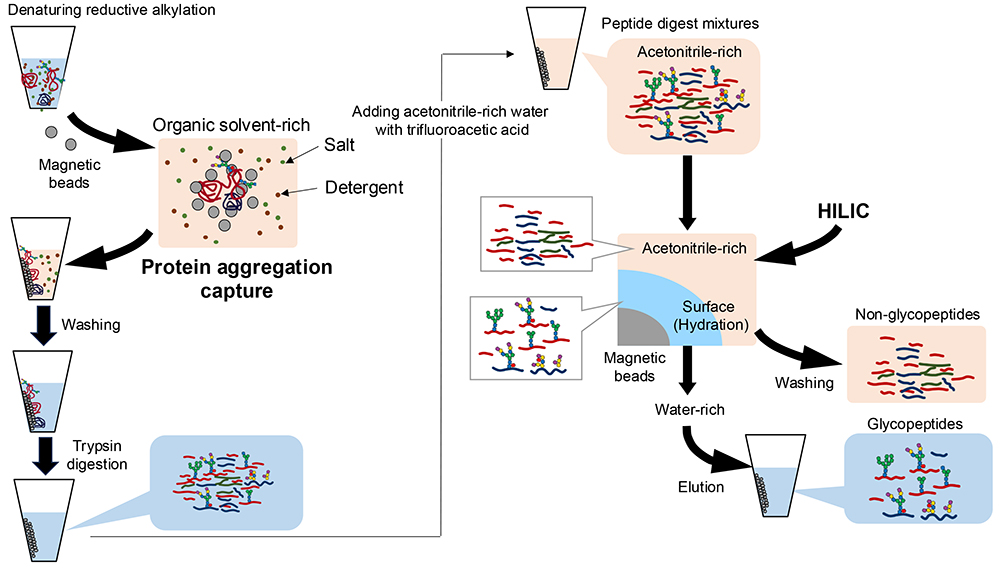
For proteomics applied to biological samples, various sample preparation strategies have been developed to enhance protein identification, ensure quantitative reproducibility, and facilitate automation. One such method, single-pot, solid-phase-enhanced sample preparation (SP3), involves sample preparation within a single tube using magnetic beads4. Notably, the SP3 method has also been adapted for glycopeptide preparation5. In glycoproteomics, the SP3 method involves peptide fragmentation (Figure 2, left) and subsequent glycopeptide purification based on a distinct principle (Figure 2, right).
The SP3 method is based on protein aggregation capture and non-selective retention of proteins aggregated onto magnetic beads in an organic solvent-rich solution (50–80% v/v ethanol, acetonitrile, etc.). The magnetic beads are then washed 2–3 times with 80% ethanol solutions to facilitate the removal of unwanted contaminants (salts, surfactants, etc.). Purified proteins are then enzymatically digested with trypsin. Moreover, with the reagent compatibility afforded by SP3, we can use detergents, such as sodium dodecyl sulfate, to solubilize a wide range of proteins. Thus, this method is applicable not only to blood samples but also to the extraction of glycoproteins from tissues for glycoproteomics analysis.
To obtain glycopeptides, digested peptide mixtures should be purified by introducing a loading solution enriched with acetonitrile into the trypsin digest (Figure 2). The purification relies on hydrophilic interaction chromatography (HILIC), which separates glycopeptides based on glycan moiety differences. In HILIC, glycopeptides are retained in the hydrated layer of the hydrophilic stationary phase in the presence of high concentrations of an organic solvent, typically acetonitrile. Non-glycosylated peptides migrate to the acetonitrile phase and can be washed away. Purified glycopeptides are then eluted in water-rich solvents.
Carboxylic acid-modified beads are commonly used for peptide fragmentation with the SP3 method. However, in glycopeptide purification, the use of stationary phase modified with polysaccharides or similar compounds improves both the recovery of glycopeptides and the non-glycopeptide removal efficiency6. Regarding the solvent content in HILIC, adding trifluoroacetic acid to the loading solution improves the non-glycopeptide removal efficiency in glycopeptide purification.
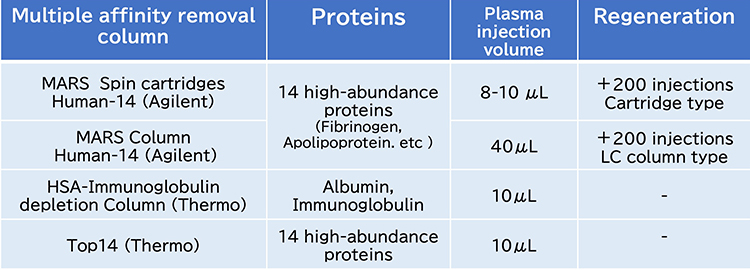
Immunodepletion using a multi-antibody column, the most common prefractionation strategy, can effectively remove 6 or 14 highly abundant proteins in blood samples (Figure 3). Agilent’s MARS columns are available in both cartridge and HPLC column formats, chosen based on the quantity of protein present. A notable advantage of these columns is their ability to be regenerated up to 200 times. However, HPLC is offline from the robotic system. Moreover, owing to the specific shapes of the cartridges, automating the regeneration process is challenging and complicates their integration into automated systems.
Albumin- and immunoglobulin-removal columns, as well as Thermo Scientific’s Top14 resins, are available in bulk, facilitating their integration into automation protocols. A comparative study in glycoproteomics revealed that approximately 2,400 glycopeptides could be identified from 10 μL of plasma using these resins. However, main obstacle with depletion strategies is co-depletion of unwanted proteins by non-specific binding. In addition, the high cost of multi-antibody columns presents a challenge. Therefore, a method that balances cost-efficiency with automation capabilities is needed to conduct large-scale cohort studies.
To ensure quantitative reproducibility across multiple samples, it is crucial to monitor chemical changes in prepared glycopeptides, such as the oxidation of methionine, which can occur after the peptides are placed into an autosampler. The oxidation of methionine makes it difficult to identify which one is correct in glycoproteomics, since Met + O + deoxyHexose = Met + Hexose.
Hydrophobic peptides and glycopeptides present in trace amounts are prone to adsorption onto the surfaces of tubes and multiwell plates during sample preparation, leading to losses of sample constituents. In proteomics, hydrophilically coated low-adsorption tubes and plates are typically used to mitigate these issues. However, the signals of hydrophobic and low-abundance glycopeptides are diminished within a few days of the sample being placed in the autosampler, through adsorption.
Peptides tend to adhere to the LC-MS system, onto trap columns, separation columns, and valves. Sample carryover is a form of cross contamination that originates from the previous analysis, and even the slightest carryover will result in false positive results in identification and quantification. To mitigate these issues, the ZebraWash procedure of alternating between elution solvent and equilibration solvent cycles in the Thermo Scientific Vanquish Neo system is used to reduce sample carryover. Additionally, an analytical blank without sample content is analyzed between glycopeptide samples to ensure data accuracy.
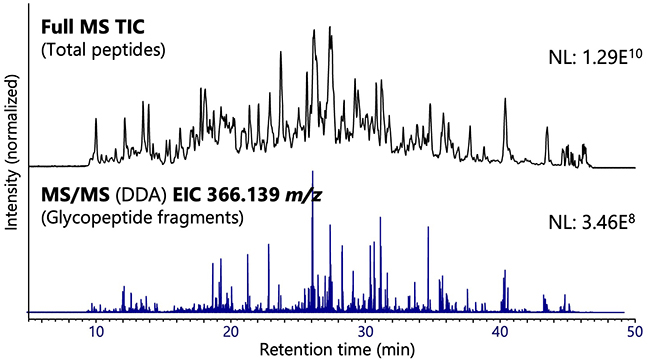
Figure 4 shows the results of analysis of glycopeptides obtained by using the SP3 method after treating the plasma (10 μL) with an albumin–immunoglobulin-removal column. The extraction ion chromatogram of fragment ions derived from lactosamine demonstrates the high purity of the recovered glycopeptides. In this experiment, 2037 glycopeptides (including 1735 sialo-glycopeptides), 497 glycosylation sites, and 173 glycoproteins were identified.
Because sample preparation is straightforward, automated LC-MS systems are increasingly used in metabolomics and blood drug concentration measurement. Typically, this involves adding an organic solvent to the blood sample, centrifuging to remove proteins, and analyzing the resulting supernatant via LC-MS. Various companies offer liquid handler products designed to automate proteomic processes. These systems feature capabilities such as liquid dispensing, agitation, magnetic bead manipulation and retrieval, temperature regulation, and barcode scanning1. They support processing capacities ranging from 8 to 384 samples per run and accommodate pipetting volumes of from 0.5 μL to several milliliters. The capacities of the deck for microplate placement and the extent of operations vary depending on the liquid handler model.
In a large-scale cohort analysis involving 200,000 HGA samples, achieving seamless and fully automated sample preparation is paramount. It is believed that currently available commercial liquid handlers might not meet these stringent requirements. Developing a fully automated system will necessitate the addition of new components, such as a centrifuge and a freezer, as depicted in Figure 5. Given the potential for errors in pretreatment devices and LC-MS systems, there is a pressing need for sample tracking in the entire system, and detailed tracking reports can be exported. Moreover, variations in blood sample quality, such as with hemolysis, can significantly affect the accuracy of opening sample containers, sample dispensing, and sample pretreatment procedures. Therefore, a fully automated device must incorporate a traceability system that extends to LC-MS measurement results, along with mechanisms to centrally oversee both sample preparation and analysis outcomes.

The Human Glycome Project (human-glycome.org)) is currently underway in the United States and Croatia. This effort involves large-scale analyses using LC-MS systems and liquid handlers imported from abroad, and it strives mainly to clarify the inter-individual variability in abundant glycoproteins as determined by targeted glycomics. On the other hand, the unique objective of the HGA in Japan is to establish a comprehensive catalog of glycopeptides in a variety of human biological samples including blood and other materials. The anticipated information output from the HGA is expected to surpass that of similar initiatives. To achieve this ambitious goal, the author believes that now is the ideal time for industry, government, and academia to collaborate in advancing the HGA agenda by leveraging Japan’s expertise in analytical equipment and robotics technology.



