
氏名:松井 佑介
2014年 北海道大学大学院情報科学研究科・博士課程修了(1年9ヶ月短縮修了)
2015年 名古屋大学医学系研究科 附属神経疾患腫瘍分子医学研究センター / リーディング大学院実世界データ循環学リーダー養成プログラム・特任助教
2018年 名古屋大学医学系研究科 総合保健学専攻ヘルスケア情報科学 生命人間情報健康医療学講座・准教授
2022年 糖鎖生命コア研究所 糖鎖ビッグデータセンター 数理解析部門・部門長(兼任)
がん、認知症、老化などの主要な疾患については、一細胞レベルも含めたゲノムワイドなマルチオミクスな分子メカニズムの知見が蓄積されているのに対し、「糖鎖」は十分に理解されていないメカニズムの一つである。これらの疾患の責任因子として作用する糖鎖とは何かについての知見には、依然として大きなギャップがある。このギャップを埋めるためには、糖鎖と他のオミックス分子情報との関係(共通性、相補性、独立性など)を明らかにすることが重要であり、そのためには統合的な解析アプローチが不可欠である。
糖鎖構造そのものは鋳型情報に直接依存しないが、その合成は糖転移酵素やそのタンパク質産物を含む糖鎖関連遺伝子に依存する。これらの糖鎖関連遺伝子は、ゲノム変異、DNAメチル化、転写調節、スプライシングなどの異常による影響を受けると考えられている。また、タンパク質配列に変異配列が含まれる場合、タンパク質に対する糖鎖の親和性も変化する可能性がある。このようなゲノミクスとグライコミクスの接点に着目することで統合解析アプローチの開発が可能となる。一方で、統合解析には大きな技術的ギャップがある。シークエンシング技術により一細胞レベルでの時空間解析は可能であるが、質量分析においては、原理的に一細胞レベルでのゲノムワイドな統合解析は容易ではない。現在の糖鎖解析技術は、糖鎖特異的抗体や糖鎖結合タンパク質(GBP、レクチンなど)を用いたプロービングに限定されており、グライコミクスのみでは一細胞分解能での生物学的な糖鎖メカニズムの解明は困難である1 。したがって、シングルセルシークエンシング技術を用いた統合解析アプローチが必要である2。
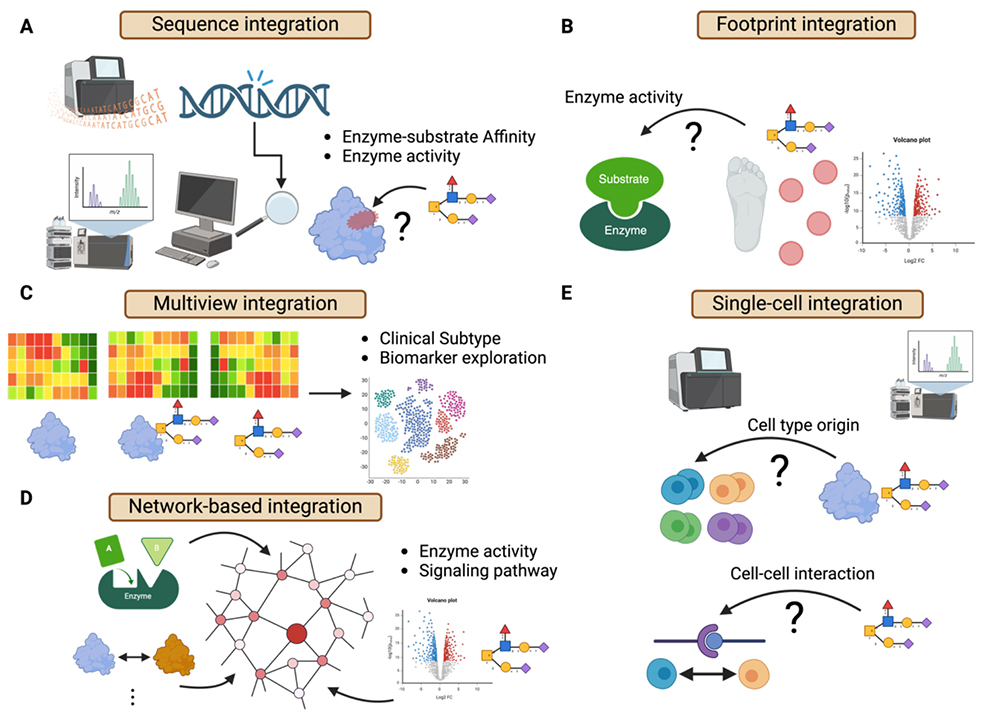
現時点において疾患マルチオミクスにおける糖鎖を用いた統合解析アプローチの研究は文献上ほとんどない。そこで、本記事ではプロテオームやリン酸化プロテオーム領域など他分野の研究を参考に、統合解析アプローチの一部を紹介する(図 1)。
BioRender.comによって作成した。
タンパク質配列内に変異がある場合、グリコシル化の過程でタンパク質に対する親和性が異なる可能性がある。プロテオゲノミクス3は、ゲノム配列に基づく統合を試みるプロテオミクスにおけるアプローチであり、ゲノムや転写レベルでの異常が、ペプチドやタンパク質レベルでの異常な変化として観察されるかを明らかにすることが可能である(図 1A)。シークエンサーベースの解析だけでは機能予測に限定されるのに対し、プロテオゲノム解析では、ペプチドレベル・タンパク質レベルにおける検証も同時に行えるため、標的探索や医薬品開発にとって魅力的である。このような計算を遂行するために、いくつかの計算手法が提案されている4,5。基本的には、インシリコでゲノム配列をアミノ酸配列に変換し、マススペクトルピークアノテーションを検索する際に参照配列に変換・組み込み、対応するペプチドを同定する方法である。このような計算フレームワークはグライコゲノミクス解析にも応用できるかもしれない。
糖鎖形成パスウェイは一般に糖転移酵素の下流にあると考えられているので、ある条件下でのグリコシル化の変化パターンは、上流の糖転移酵素活性の変動を反映している可能性がある。言い換えれば、グライコミクスやグライコプロテオミクスで得られる測定値の変動は、糖転移酵素の「足跡」を観察していると考えられる。ここでの問題は、フットプリントから上流の酵素活性をどのように推測するかである(図 1B)。フットプリント解析のアプローチは、リン酸化プロテオミクスの分野で比較的よく研究されている6。
リン酸化プロテオミクスでは、まず各キナーゼとそれらが標的とする基質部位との対応関係を蓄積したデータベースが構築されている7。それらを用いて、条件間で存在量が変化するリン酸化部位から上流のキナーゼ活性を推測する。例えば、Kinase Enrichment Analysis (KEA)8,9やKinase Substrate Enrichment Analysis (KSEA)10などが代表的な手法である。残念ながらグライコミクス分野では、このような包括的アプローチに基づくデータベースは存在しないため、KEAのような解析は今のところ不可能である。しかし、糖鎖構造と基質を対応付けた包括的なデータベースが構築できれば、同様のアプローチが可能になる。
1回の実験で多くの分子パラメータを測定する多層オミックス解析研究の利点は、複雑な疾患メカニズムの「多面的解析」を可能にし、「階層間の複雑な相互作用」にアプローチできることである11。たとえば、コホートデータのような集団レベルに対して、プロテオーム、グライコプロテオーム、グライコームを同時取得した場合には、タンパク質、糖タンパク質あるいは糖鎖のそれぞれに対して疾患表現型と関連性に関わる情報が得られるだけでなく、たんぱく質―糖鎖、糖転移酵素―糖鎖のような階層間の相互作用と疾患表現型との関連性についての洞察も得られる。また、複数の階層にまたがるバイオマーカーを探索し、分子プロファイルと臨床情報によって特徴付けられる疾患サブタイプを絞り込むことによって、診断予測の精度を向上させることも可能である。
マルチビュー学習12,13は、多階層の統合を可能にする機械学習のフレームワークの一つである(図 1C)。このアルゴリズムのフレームワークは、各階層(シングルビュー)レベルの特徴量だけでは得られない、複雑な階層間(ビュー間)の相互作用に由来する潜在的なパターン(サンプルのサブグループ構造など)を学習し、分類や予測を行う点に特徴がある。元々は、ビデオ、オーディオ、テキストなど様々なデータタイプのマルチモーダル統合解析のために開発されたものであるが、がんや神経疾患のマルチオミクス解析の文脈においても応用されている14。
分子生物学のメカニズムには、複数の分子が協調して機能を発現していると考えられている。このような協調に関する知識は、ネットワーク科学の言語によって抽象化されることが多い。様々な既知の相互作用を埋め込んだネットワークを利用することで、機能が未知の分子や基質が関与する生物学的プロセスを予測する方法を構築することができる(図 1D)。
リン酸化プロテオミクスでは、ネットワーク科学のアプローチがキナーゼ活性の推定に有効であることが示されている。例えば、ROKAI法は15は、1)PhosphositePlus7を用いたキナーゼ-基質ネットワーク2)PTMcode16を用いたリン酸化部位間の共進化と構造的距離を表すネットワーク 3)STRING17を用いた統合されたキナーゼ相互作用ネットワークを統合したネットワークを最初に構築する。さらに、このネットワーク上でリン酸化部位の定量値を伝播させ、定量値が一貫して変化するリン酸化部位をスコアリングすることでキナーゼ活性を予測する。このように異種の相互作用ネットワーク(糖鎖遺伝子の転写ネットワーク、タンパク質相互作用、糖転移酵素の酵素-基質相互作用など)とグライコミクス・グライコプロテオミクスからの定量値を組み合わせることで、既存の生物学的知見と整合性のある予測が可能になるかもしれない。
シングルセルシークエンシング技術は、細胞組織の構成、細胞の発生段階における動的プロセス、あるいは病理学や治療に対する反応における転写不均一性や細胞間相互作用の役割を明らかにするための強力なツールであり、大規模なアトラスが日常的に発表されている。これらの蓄積された一細胞プロファイルを利用して、細胞型の注釈を付与する統合的解析も広く利用されている18。例えば、バルクRNAシーケンスやプロテオミクスから得られた特異的分子や変動分子をクエリーとして用いて、濃縮解析に基づいて細胞型アノテーションを付与することができる。
このような方法を用いれば、特定の疾患に関与する糖鎖遺伝子あるいはタンパク質の細胞型起源を同定することが可能である。さらに、糖鎖は細胞表面に発現しており、細胞間相互作用において重要な役割を担っている可能性が高い(図 1E)。シングルセルシークエンシング解析は、細胞型ごとのリガンドおよび受容体の発現プロファイルに基づいて、細胞間相互作用を予測することができる。このようにして得られる細胞間相互作用の情報と糖鎖情報を統合し、細胞間相互作用に関与する糖鎖修飾因子をゲノムワイドに同定するアプローチも、今後可能になるかもしれない。
本稿では、疾患特異的な糖鎖マルチオミクス研究を念頭に、統合解析アプローチの概要を紹介した。我々の知る限り、糖鎖特異的な疾患メカニズムにアプローチするために、複数のオミックス階層を積極的に統合した研究はほとんどない。重要なことは、単に多層的なマルチオミクス情報を得るだけでは不十分な場合もあるということである。例えば、リン酸化プロテオミクスの例で紹介したように、リン酸化プロファイルから上流のキナーゼ活性を推定する方法を開発するためには、ゲノムワイドな酵素と基質の組み合わせに関するデータの知識が得られていなければならない。 したがって統合解析を見据えた知識ベース開発も重要な研究課題である。最後にもう一つ強調しておきたいのは、他の分野で既に研究されている計算アプローチを転用することが可能である点である。オミクス研究の強みの一つは、オープンサイエンスの文化が根付いていることであり、転用可能なオープンソースの解析ツールが数多く存在している。これらを適切に取り入れることで、疾患理解における新たな次元としての「糖鎖」を解明する強力なツールになると考える。



