
Yusuke Matsui
2014 - Completed Ph.D. program in Information Science at Hokkaido University, Japan, finishing 1 year and 9 months ahead of schedule.
2015 - Designated Assistant Professor at the Center for Neurological Diseases and Cancer, Graduate School of Medicine, Nagoya University / Leader of the Real World Data Circulation Leaders, Program for Leading Graduate School Nagoya University.
2018 - Associate Professor at the Biomedical and Health Informatics Unit (Principal Investigator), Department of Integrated Health Science, Graduate School of Medicine, Nagoya University.
2022 - Head of the Systems Biology Division at the Integrated Glycan-Big Data Center (iGDATA), Institute for Glyco-core Research (iGCORE), Tokai National Higher Education and Research System.
Whereas for major diseases such as cancer, dementia, and aging, we have accumulated knowledge of genome-wide, multi-omics molecular mechanisms, including at the single cell level, the mechanism of glycan-mediated biological processes is not well understood. There remains a significant gap in our knowledge of the glycans responsible for these diseases. To fill this gap, it is important to clarify the relationship between glycans and other omics molecular information, such as commonality, complementarity, and independence, for which an integrated analysis approach is essential.
Although glycan structures themselves do not directly depend on template information, their synthesis depends on glycan-related genes, including glycosyltransferases and their protein products. These glycan-related genes are thought to be affected by genomic mutations, DNA methylation, transcription factors, and splicing defects. The susceptibility of proteins to glycosylation may also be altered if the protein sequence contains mutated sequences. However, there are significant technical gaps in the integrated analysis. Although sequencing allows spatiotemporal analysis at the single-cell level, genome-wide integrated analysis at the single-cell level is feasible but not easy to achieve using mass spectrometry. Current glycan analysis technology is limited to probing with glycan-specific antibodies and glycan-binding proteins (GBPs) (lectin, etc.), making it difficult to elucidate biological glycan formation mechanisms at the single-cell level using glycomics alone1. Therefore, integrated analysis using single-cell sequencing technology is necessary to understand the mechanism of abnormal glycosylation at the single-cell level2.
Presently, the literature on disease-specific glycan multi-omics contains few integration analysis approach studies. Therefore, we present some of the integrated analysis approaches with reference to studies in other fields such as proteomics and phosphoproteomics (Figure 1).
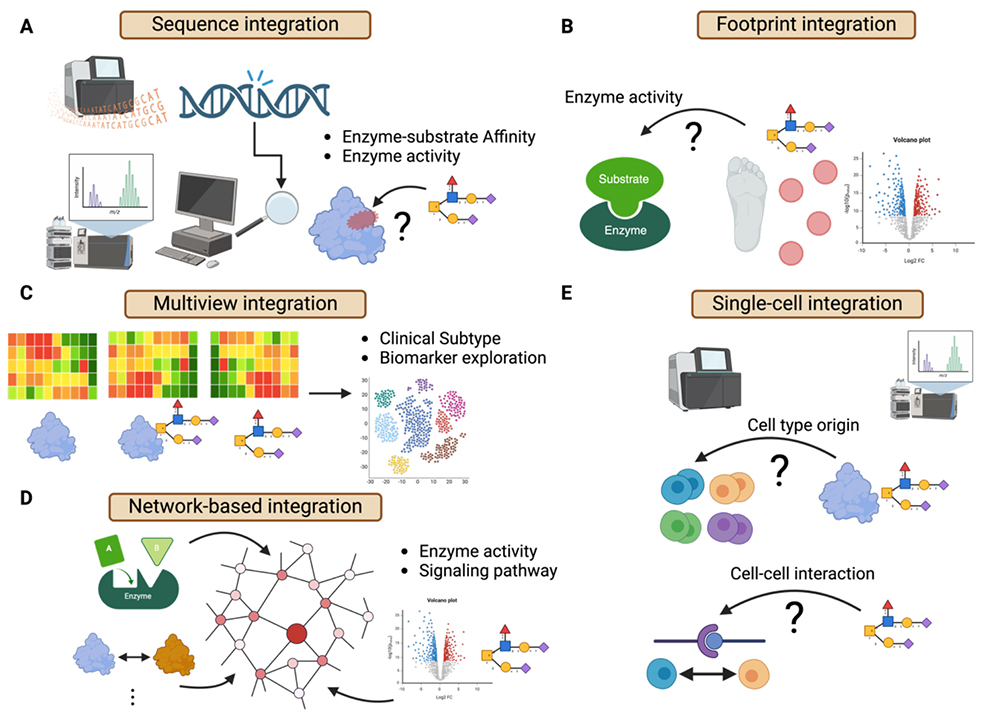
Figure created with BioRender.com.
The process of glycosylation may also have a different affinity for proteins if mutations are present within the protein sequence. Proteogenomics is a proteomics approach3 that attempts sequence-based integration. It is possible to reveal abnormalities at the genomic or transcriptional level that are observed as aberrant changes at the peptide or protein level (Figure 1A). Whereas sequencer-based analysis alone is limited to functional prediction, integrated analysis can provide validation at the peptide and protein levels simultaneously, which is attractive for target discovery and drug development. Several computational methods have been proposed to achieve this purpose4,5. All of these methods convert the genome sequence into an amino acid sequence in silico, and when searching for mass spectral peak annotations, the genome sequences are transformed and incorporated into a reference sequence, thereby identifying the corresponding peptides. These computational frameworks may also be applicable in glycogenomics analysis.
Because glycosylation pathways are typically considered to be downstream of glycosyltransferases, the pattern of glycosylation changes under certain conditions may represent variations in upstream glycosyltransferase activity. In other words, fluctuations in measurements obtained by glycomics or glycoproteomics are considered to be the "footprints" of glycosyltransferases. The question is how to infer upstream enzyme activity from the footprint (Figure 1B). The footprint analysis approach has been relatively well-studied in the field of phosphoproteomics6.
In phosphoproteomics, an established approach is to first build a database that accumulates the correspondence between each kinase and the substrate sites they target7 and then use it to infer upstream kinase activity from phosphorylation sites that change in abundance between conditions. Kinase Enrichment Analysis (KEA)8,9 and Kinase-Substrate Enrichment Analysis (KSEA)10 are among the leading methods. Unfortunately, data based on such a comprehensive approach do not exist in the field of glycomics, and thus, KEA-like analysis is currently not possible. However, construction of a comprehensive database that maps glycan structures to substrates would make a similar approach feasible.
Multilayered omics analysis studies, in which many molecular parameters are measured in a single experiment, have the advantage of allowing multifaceted analysis of complex disease mechanisms and complex interactions between layers11. For example, at the level of population studies, such as cohort studies, the simultaneous collection of proteome, glycoproteome, and glycome data provides not only data about proteins, glycoproteins, and glycans that are relevant to disease phenotypes and their association but also insights into the interaction between different molecular layers, such as protein-glycans and glycosyltransferase-glycans, that are relevant to disease phenotypes. It is also possible to improve the accuracy of diagnostic prediction by exploring biomarkers across multiple layers and refining the disease subtypes characterized by molecular profiles and clinical information.
Multi-view learning12,13 is one framework of machine learning that allows for multi-layer integration (Figure 1C). For purposes of classification and prediction, this algorithmic framework is unique in that it learns latent patterns (e.g., sample subgroup structure) derived from complex layered (inter-view) interactions, which cannot be obtained from features at each (single-view) level alone. Originally developed for the integrated analysis of various data types, such as video, audio, and text, several methods have been proposed and applied to multi-omics analysis of cancer and neurological diseases14.
Molecular biological mechanisms are thought to involve multiple molecules working in concert to express their functions. Knowledge of such coordination is often abstracted from the language of network science. By utilizing networks that embed various known interactions, it is possible to develop methods for predicting biological processes involving molecules and substrates whose functions may be unknown (Figure 1D).
In phosphoproteomics, network science approaches have also been shown to be effective for inferring kinase activity. For example, the ROKAI method15 uses (1) a kinase-substrate network with the PhosphoSitePlus database716, (3) integrated kinase-interaction network with the STRING database17. Changes in kinase activity can be predicted by generating quantitative values of phosphorylation sites in this network and scoring groups of phosphorylation sites with consistent changes in quantitative values. This combination of heterogeneous interaction networks (transcriptional networks of glycogenes, protein interactions, and enzyme-substrate interactions of glycosyltransferases) and quantitative values from glycomics or glycoproteomics allows predictions to be consistent with existing biological knowledge.
Single-cell sequencing technology is a powerful tool for revealing cellular tissue composition, dynamic processes during cell developmental stages, or the role of transcriptional heterogeneity and cell-cell interactions in pathology and response to therapy, and large atlases are routinely published. Integrative analyses that take advantage of these accumulated single-cell profiles to assign cell type annotations are also widely used18. For example, enrichment analysis methods use bulk RNA sequencing or proteomics data on specific or variable molecules to assign cell type annotation.
Using such methods, it is possible to identify the cellular origin of glycogenes involved in specific diseases. In addition, glycans are expressed on the cell surface and likely have an essential role in cell-cell interactions (Figure 1E). Single-cell sequencing analysis can predict cell-cell interactions based on ligand and receptor expression profiles for each cell type. Such an approach, which integrates cell-cell interaction information with glycan multi-omics profiling information to identify responsible glycosylation factors for cell-cell interactions on a genome-wide basis, is expected to become important in the future.
In this article, an overview of integrated analysis approaches is presented, with disease-specific glycan multi-omics studies in mind. To the best of our knowledge, few studies have actively integrated multiple omics layers to approach glycan-specific disease mechanisms. Importantly, simply acquiring multilayered multi-omics information is insufficient. For example, as introduced in the example of phosphoproteomics, it is only when a priori knowledge of data pertaining to genome-wide combinations of enzyme-substrate is obtained that it is possible to develop methods for inferring upstream kinase activity from phosphorylation profiles.
Finally, it is important to emphasize that it is possible to use the approaches that have been studied in other fields. There are also a number of computational tools that can be adapted. By combining these approaches with those of existing disease multi-omics, it will be possible to clarify the role of glycans.



