
Takashi Hosoya
Associate professor at grduate school of life and environmental sciences, Kyoto prefectural university.
The author graduated from the Faculty of Agriculture, Kyoto University in March 2003, and completed a doctoral program at the graduate school of energy science, Kyoto university in May 2008. Subsequently, the author served as a Japan society for the promotion of science research fellow at Kyoto university and a postdoctoral researcher at the university of natural resources and life sciences, vienna. The author then held positions as a specially appointed lecturer and assistant professor at the graduate school of life and environmental sciences, Kyoto prefectural university. Since October 2019, the author has been in the current position. The author specializes in biomass chemistry and physical organic chemistry.
The chemical synthesis of glycosidic bonds (glycosylation) is a key reaction enabling the artificial synthesis of various glycoconjugates and polysaccharides. Glycosylation involves a nucleophilic substitution process between a glycosyl donor, in which various leaving groups are introduced at the anomeric carbon of a sugar, and an acceptor molecule. Nucleophilic substitution reactions are elementary organic chemistry reactions encountered at an introductory university level, and it might seem that established methods for controlling them exist. However, this reaction progresses through a complex process, distinct from typical nucleophilic substitutions, wherein the electrophilic donor forms an equilibrium mixture composed of various constituents. Due to this complexity, the control of glycosylation reactions often relies on the trial and error based on the synthetic chemist’s experience and intuition. To overcome this situation, elucidation of the molecular mechanisms involved in this reaction has been pursued by many researchers. This paper focuses on evaluating the chemical properties of ion pairs, which are crucial in the elucidation of the reaction mechanisms of glycosylation, and introduces various studies in this field.
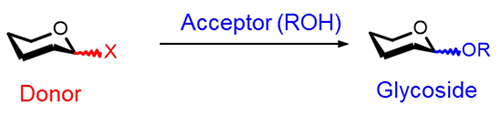
The chemical synthesis of glycosidic bonds, namely organic synthetic glycosylation, represents one of the primary research areas within carbohydrate chemistry. This reaction is a nucleophilic substitution involving a glycosyl donor bearing a leaving group X and acceptors such as alcohols, amines, or olefins, as depicted in Figure 11-5. Particularly, glycosylation reactions employing alcohols as acceptors (O-glycosylation) are indispensable for the chemical synthesis of various glycoconjugates, oligosaccharides, and polysaccharides, and hold a critically important position not only in organic chemistry but also in biochemistry and medicine. In the field of wood chemistry, which is the author's specialty, the synthesis of various model sugars via glucosylation is conducted to systematically investigate the properties of cellulose and hemicellulose, major components of wood, from a chemical structural perspective6,7.
Despite the critical importance of glycosylation, this reaction cannot yet be considered a well-established reaction from the perspective of organic synthetic chemistry. The success or failure of synthesizing a desired sugar molecule envisioned by a synthetic chemist often depends on the chemist's experience, intuition, and notably, the extent of effort expended. Among the challenges this reaction faces, stereoselectivity (α/β selectivity) is particularly significant. To establish the synthesis conditions for a single sugar molecule, meticulous attention is required to set a wide range of reaction parameters, from relatively simple factors like temperature and solvent, to more complex aspects such as the nature of the leaving group X and the protective groups on donor’s hydroxy groups, in order to suppress the formation of unwanted isomers. The difficulty inherent in this process can be appreciated from the fact that while synthetic methods for proteins and nucleic acids are well-established, the establishment of synthetic methods for oligosaccharides and polysaccharides remains unachieved.
Although glycosylation reactions are fraught with many challenges, a contributing factor to the current situation is the lack of mechanistic insights that should serve as a basis for rationally examining various synthetic conditions. The nucleophilic substitution reaction depicted in Figure 1 is a topic of introductory-level university organic chemistry, yet this representation alone does not suffice to understand the essence of the reaction. To base glycosylation control methods on reaction mechanisms, more detailed mechanistic insights from a microscopic perspective are necessary. These insights are crucial for developing rational strategies that could effectively manipulate the complex conditions governing the synthesis of glycosidic bonds.
Figure 2 illustrates the glycosylation mechanism widely studied today, utilizing a donor molecule with a triflate (trifluoromethanesulfonate, TfO) group as the leaving group X 5. The compound where C-1 and TfO are linked by a covalent bond is termed a covalent triflate (CT). However, it is believed that CT represents only a small fraction of the true nucleophilic species present in the reaction system. The C-1—OTf bond in CT undergoes spontaneous ionic dissociation even in nonpolar solvents commonly used as reaction media in glycosylation, such as dichloromethane. This dissociation is driven not only by the high stability of the TfO– ion but also by the significant stabilization of the resulting oxacarbenium ion at the C-1 cationic center through charge transfer from O-58. As the ionic dissociation of the covalent bond between C-1 and TfO progresses, a contact ion pair (CIP) forms, where the CIP processes loose electrostatic interactions between the cation and anionic parts. Further ionic dissociation from CIP leads to the formation of a solvent-separated ion pair (SSIP). Given the sufficiently low activation barriers between CT, CIP, and SSIP transitions, glycosyl triflates rapidly form an equilibrium mixture consisting of CT and various ion pair species upon formation in the reaction system.
CT, CIP, and SSIP, each depicted in Figure 2 as reacting with the acceptor designated as ROH, yield glycosides through different reaction modes corresponding to the nature of each electrophilic species. The covalently bound CT reacts with the acceptor through an Sɴ2 mechanism involving stereochemical inversion at C-1. This means that from αCT (α-configurated CT, shown in Figure 2), β-glycosides are formed, whereas from βCT, α-glycosides are produced. On the other hand, the reaction involving SSIP is thought to proceed via an Sɴ1-like mechanism with the acceptor, resulting in the formation of both α- and β-glycosides. The nucleophilic substitution reaction of CIP occurs through a mechanism intermediate between those of CT and SSIP. Generally, it is understood that this involves an Sɴ2-like reaction where the acceptor attacks the donor molecule from the side opposite to that where TfO– is located.
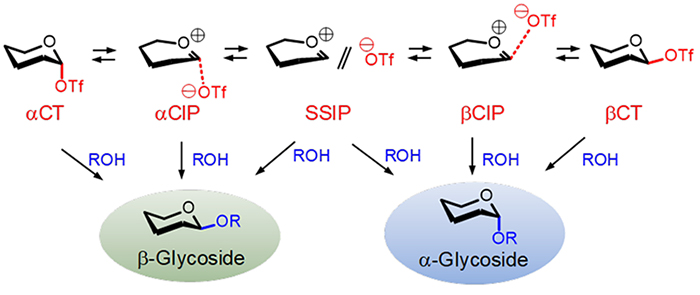
In glycosylation reactions, the α/β selectivity of the products is determined by the relative rates of the nucleophilic substitution reactions proceeding from CT and the various ion pairs. Thus, from the perspective of controlling α/β selectivity, it is crucial to systematically understand the chemical characteristics of CT and ion pairs (such as their chemical structures and relative energies) in relation to their reactivity with the acceptor. However, within the equilibrium mixtures depicted in Figure 2, the chemically detectable species at low temperatures, such as through NMR, are predominantly αCT in many cases9. This predominance is primarily due to the stabilization of αCT by the anomeric effect within the mixture in Figure 2.
Currently, it is extremely difficult to obtain detailed information on chemical species other than αCT. This is evident not only from the substantial number of reports on structural analyses of αCT using low-temperature nuclear magnetic resonance (NMR) methods9-11 but also from the fact that there are few documented instances of detecting βCT, another covalent species12. Moreover, there are hardly any reports on the detection of ion pairs, which are considered to be less stable and short-lived in the reaction mixture, with only a few exceptions13. However, even in reaction systems where only αCT is detected using low-temperature NMR, the actual glycosylation reactions usually produce some α-glycosides with retained stereochemistry at C-1. This experimental fact serves as one piece of evidence supporting the existence of the equilibria depicted in Figure 2.
The difficulty in detecting and characterizing the actual chemical species involved in nucleophilic substitution reactions with acceptors is a major factor complicating the full elucidation of the reaction mechanisms in glycosylation. In response to this issue, information about the reactivity of CT and ion pairs, which are components of the equilibrium mixtures depicted in Figure 2 in solution, is being studied using indirect methods. For instance, investigating the isotopic effect at C-1 on the α/β ratio of the produced glycosides can yield valuable insights into the contribution of ion pairs in the glycosylation reaction14. There are also reports evaluating the Sɴ2 and Sɴ1 character in nucleophilic substitution reactions based on the glycosylation behavior of special donor molecules that induce neighboring group participation at C-115. On the other hand, direct detection of ionic species has been attempted through analyses using mass spectrometry or infrared spectroscopy to study oxacarbenium ions16,17. However, it is important to note that such direct analyses are often conducted in high vacuum conditions within analytical equipment, which may provide information that differs from what occurs in solution during actual glycosylation processes.
First-principles calculations and density functional theory (DFT) have become commonplace for interpreting and predicting various chemical phenomena among contemporary chemists. Notably, DFT calculations are employed to compute unstable intermediates and transition states in the glycosylation mechanism18-24. Ion pairs, as depicted in Figure 2, are typical unstable chemical species, and their evaluation seems well-suited to theoretical computations. However, even with recent advances in theoretical chemistry, accurately calculating the process of ion pair formation from CT remains a considerable challenge. For example, extending the C-1—OTf bond in glycosyl triflates and calculating the potential energy curve related to the glycosidic bond length using methods like MP2 or DFT often results in an energy profile that shows a monotonic increase with bond distance, and does not predict CIP as a stable structure directly originating from CT. Furthermore, starting structural optimization calculations with the oxacarbenium ion and TfO– at sufficiently separated distances and in various relative positions fails to yield ion pair structures; instead, the structures that emerge result in either α or βCT structures reflecting the initial relative arrangements of the cation and anion. This tendency persists even when solvent effects are incorporated into the calculations using frequently utilized solvation models such as polarizable continuum models.
In essence, the problem can be stated as follows: as long as the computational schemes commonly used in computational reaction analysis are employed, the ion pairs depicted in Figure 2 are computationally non-existent, and therefore, their properties cannot be computed. While the Sɴ2 nucleophilic reactions between the stable structure CT and various acceptors can be relatively easily calculated, accurately predicting the experimental α/β ratio from the activation barriers of the computed Sɴ2 reactions forming α/β-glycosides from α/βCT is generally challenging and often imprecise. Several factors could be contributing to this issue, including the limitations of the computational methods employed—often DFT calculations, which are convenient but lack quantitative accuracy. Additionally, a significant underlying cause is likely the omission of various nucleophilic reactions that should be proceeding from CIPs and SSIPs, which are not even considered in most computational analyses.
A potential solution to the challenge of computing ion pairs was identified in pioneering research by Whitefield and colleagues24. They reported that by coordinating Li+ with the TfO group and conducting structural optimization, it is possible to stabilize the structure of CIPs, which are thought to exhibit mild electrostatic interactions between the oxacarbenium ion and TfO–. The success of this approach is believed to stem from the Li+ cation counteracting the negative charge accumulating on the TfO group as a result of ionic dissociation from the C-1—OTf bond. Their DFT calculations optimized the structure of a CIP possessing a half-chair pyranosyl ring conformation known as 4H3, following the dissociation of the αCT's C-1—OTf covalent bond. However, this computational method does introduce the challenge of artificially including Li+, which does not actually exist in the reaction system, into the computational model.
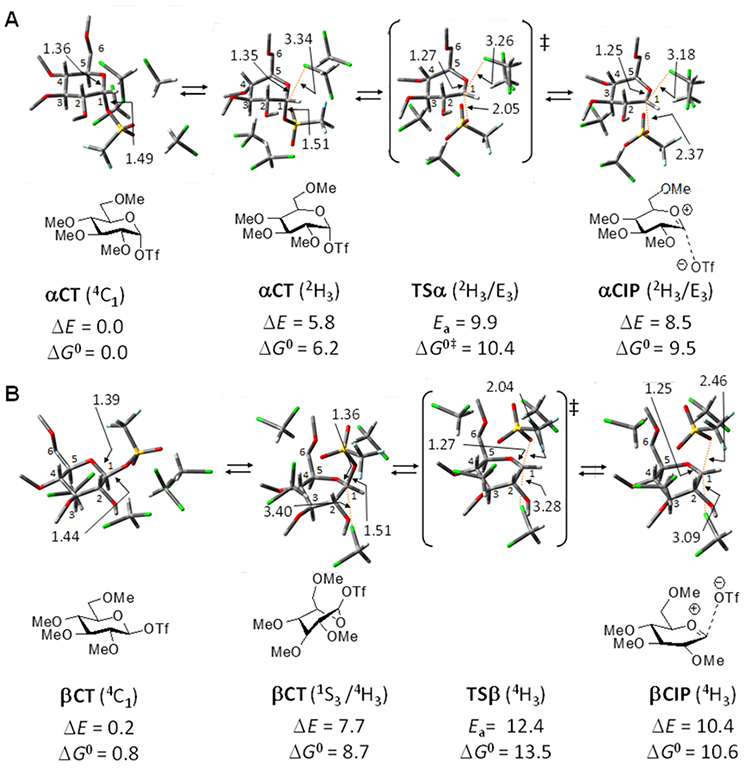
The author has also employed computational methods to evaluate the structure and stability of ion pairs in a series of glycosylations involving triflate (TfO) as the leaving group. When investigating the ionic dissociation of α-glucosyl triflate αCT, which possesses methyl group as a model protective group as shown in Figure 3A, in dichloromethane, it was found that positioning four solvent molecules explicitly on the α/β face of the donor allows the ion pairs to be computed as stable species without the need for placing Li+. Specifically, calculations using the MP4(SDQ) method and DFT(M06-2X) predicted that the formation of αCIP from αCT involves a transition from the most stable chair conformation (4C1) to a half-chair (2H3), followed by a transition state TSα leading to the formation of αCIP with a 2H3/E3 conformation. Resulting αCIP is 8.5 kcal/mol less stable on a potential energy basis (9.5 kcal/mol on a Gibbs energy basis) than αCT(4C1), corresponding well with the lack of ion pair detection in low-temperature NMR experiments. In the case of βCT, the formation of βCIP results in a different conformation (4H3) than the α-variant, and βCIP is found to be 1 – 1.5 kcal/mol less stable than αCIP (ΔE=10.4 kcal/mol, ΔG=10.6 kcal/mol, Figure 3B). Furthermore, various configurations of TfO– and solvent molecules have been optimized for SSIPs. SSIPs tend to adopt the 4H3 conformation preferred by the independent glucosyl oxacarbenium ions, and exhibit energy levels comparable to those of the CIPs.
Similar approaches involving explicit handling of solvent molecules for evaluating ion pairs have been applied to a mannosyl donor23. In this study, while ion pairs derived from glucosyl donors tend to prefer half-chair conformations like 4H3, it has been reported that ion pairs from mannosyl donors favor boat-like conformations such as B2,5. Additionally, computations have been conducted on the effect of a 4,6-di-acetal protecting group, which has been reported to facilitate the synthesis of synthetically challenging β-mannosides and other 1,2-cis glycosides22. Calculations at the DFT(M06-2X) level for a mannosyl triflate incorporating the 4,6-di-acetal structure suggest that this modification enhances the relative stability of αCIP over other ion pairs, thereby enabling the selective synthesis of β-mannosides. These findings align well with experimental mechanistic analyses using isotopic effects at C-114,25, suggesting that a combined approach of computational and experimental studies could further elucidate the complete mechanism of glycosylation reactions. This promising integration of computational predictions and empirical data points to a robust methodology for uncovering detailed insights into glycosylation processes, thereby driving advancements in synthetic carbohydrate chemistry.
In this column, the author has discussed the current state and challenges in elucidating the reaction mechanisms involved in glycosylation. It is hoped that even a simple nucleophilic substitution reaction as depicted in Figure 1 reveals the profound chemistry underlying it. While the prospect of chemists being able to freely create desired carbohydrate molecules seems distant, achieving this will necessitate unraveling the glycosylation mechanism with the precision described in this column for effective reaction control.
The author has just begun to expand research on glycosylation mechanisms, focusing on computational chemistry. Computational methods are proving invaluable for assessing structures and energies of unstable intermediates and transition states, and are indeed essential for evaluating ion pairs, which are inherently unstable intermediates. Meanwhile, recent developments in experimental techniques are also impressive. For instance, reports have used a special type of NMR analysis known as exchange NMR to study ion pairs13. Future research in this field is expected to advance our understanding of glycosylation mechanisms through closer integration of experimental and theoretical studies, driving significant progress in the chemical synthesis of complex carbohydrates.



