
Motoi Kanagawa
Professor, Department of Cell Biology and Molecular Medicine, Ehime University Graduate School of Medicine
In 2001, he graduated with a Ph.D. from the Division of Chemistry, Graduate School of Science, Hokkaido University (under Prof. Kazuya Taniguchi). Since 2001, he has been engaged in functional analysis of dystroglycan under Dr. Kevin P. Campbell at Howard Hughes Medical Institute/The University of Iowa, and has been involved in the discovery of glycosylation-defective muscular dystrophy. Since 2006, he has been engaged in research on the structure and function of dystroglycan, the elucidation of the pathogenesis of muscular dystrophy, and the development of muscular dystrophy treatments under Professor Tatsushi Toda (Osaka University/Kobe University, at present The University of Tokyo). He has been in his current position since 2020 and has been working to elucidate the pathogenesis of muscular dystrophy and develop its treatment.
Ribitol phosphate is a sugar alcohol phosphate, which is known as a component of teichoic acid in bacterial cell walls. In 2016, ribitol phosphate was found in vertebrate cells as a sugar chain component of dystroglycan. At the same time, a group of enzymes involved in the biosynthesis of ribitol phosphate was also identified, and mutations in the genes encoding these enzymes are responsible for several types of muscular dystrophy. As the mechanism of ribitol phosphate modification is revealed, the development of therapies for ribitol phosphate-defective muscular dystrophy is heating up. This article describes the history of ribitol phosphate discovery and the therapeutic strategies currently proposed.
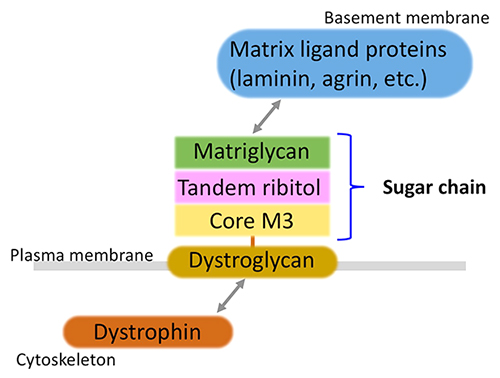
Muscular dystrophy is a group of inherited disorders with progressive muscle weakness. Currently, more than 50 genes, whose mutations are associated with muscular dystrophy, have been identified. Several genes encode structural proteins, such as dystrophin, which is the causative protein for Duchenne/Becker type muscular dystrophy. In the early 2000s, a group of muscular dystrophies caused by abnormal glycosylation of a membrane protein called dystroglycan was discovered, establishing the new disease concept of dystroglycanopathy1. Dystroglycan binds to dystrophin intracellularly, while it binds to basement membrane components such as laminin extracellularly via sugar chains (Fig. 1). Abnormal glycosylation of dystroglycan results in loss of matrix-binding ability, leading to the development of the disease.
Matrix ligand binding activity requires an O-mannose-type glycan, which was discovered by Dr. Tamao Endo's group2. This was discovered in the late 1990s, and it took a long time until the structural details of ligand binding glycan were clarified. In the 2010s, with the development of genome analysis technology, the causative genes of dystroglycanopathy were identified one after another, and the glycan structures were also gradually clarified each time. It was revealed that a repeating disaccharide unit consisting of glucuronic acid and xylose (termed matriglycan) serves as the ligand-binding domain3, and that matriglycan is modified at the end of an O-mannose-type sugar chain called Core M34 (Fig. 2). On the other hand, it was also suggested that an unknown structure exists between matriglycan and Core M3.
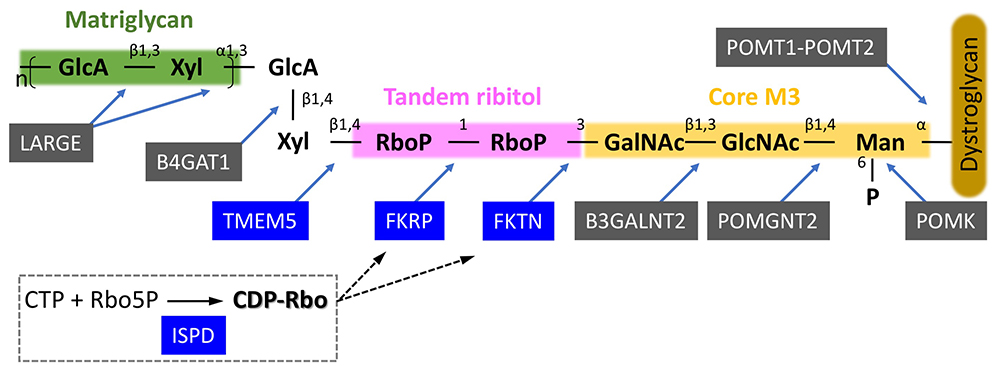
We generated matriglycan-deficient recombinant dystroglycan based on genetic information from a patient with dystroglycanopathy. By using various glyco-chemical technologies, such as glycopeptide mass spectrometry, accurate mass spectrometry, and gas chromatography/mass spectrometry, we found that two ribitol phosphates are present in the sugar chain as a tandem structure5. The dystroglycanopathy genes with unknown functions at this point were FKTN (fukutin), FKRP (FKTN-related protein), ISPD (isoprenoid synthase domain-containing protein), and TMEM5 (transmembrane protein 5). We conducted functional analyses of these 4 genes with reference to the biosynthetic pathway of bacterial teichoic acid, and found that ISPD is an enzyme that synthesizes CDP-ribitol (a donor for ribitol phosphate); FKTN and FKRP are enzymes that sequentially transfer ribitol phosphate to the Core M3 structure using CDP-ribitol as a donor substrate; TMEM5 is a xylose transferase that builds a priming structure for matriglycan synthesis on tandem ribitol phosphate5,6 (Fig. 2). FKTN is the causative gene for Fukuyama muscular dystrophy, which accounts for the majority of dystroglycanopathy patients in Japan7. FKRP is the causative gene for limb-girdle muscular dystrophy 2I, the most common dystroglycanopathy in Europe and the U.S. ISPD mutations have also been reported in more than a small number of cases from Europe, the U.S., and China. Based on the above, defects in ribitol phosphate modification are the cause of dystroglycanopathy in the majority cases.
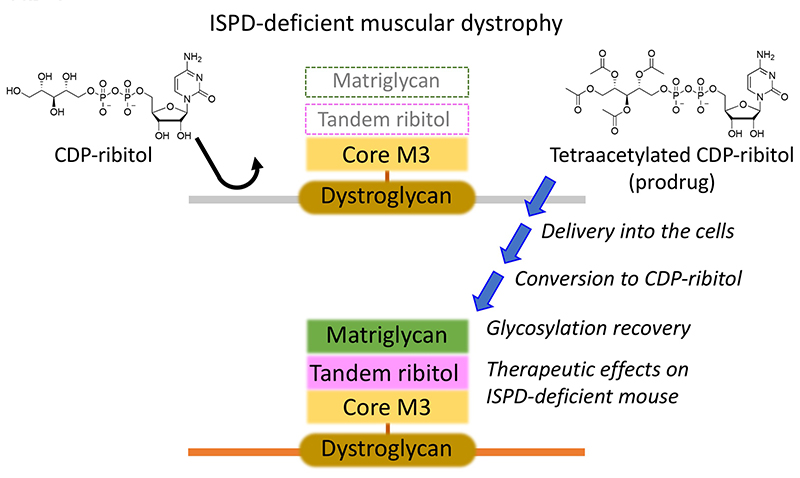
Because ISPD-deficient muscular dystrophy is caused by defective synthesis of CDP-ribitol, supplementation of CDP-ribitol from extracellular sources is expected to restore glycosylation and have a therapeutic effect5. However, CDP-ribitol is highly hydrophilic and its membrane permeability is expected to be low (Fig. 3). Therefore, we developed CDP-ribitol derivatives as prodrugs to examine the efficacy of CDP-ribitol supplementation therapy. To do this, the hydrophilic groups on CDP-ribitol were modified with hydrophobic groups to increase membrane permeability and these derivatives can be converted back to the original CDP-ribitol through cellular metabolism after intracellular delivery. We generated 10 prodrug compounds, evaluated their activity on glycosylation recovery and toxicity both in vitro and in vivo, and then selected tetraacetylated CDP-ribitol as the compound with the best prodrug activity. We found that long-term intramuscular administration of tetraacetylated CDP-ribitol significantly ameliorated muscular dystrophy symptoms in Ispd-deficient mice8 (Fig. 3). While advocating for CDP-ribitol supplementation therapy, we acknowledge much room for improvement in intracellular delivery efficiency and in vivo stability to develop systemic administration in the future.
CDP-ribitol is synthesized from ribitol 5-phosphate, but it is unrevealed how ribitol 5-phosphate is biosynthesized. Because administration of ribitol or ribose in normal cells or normal mouse muscle tissue increased the synthesis of CDP-ribitol9, it is thought that the pentose phosphate pathway is involved in the biosynthesis of CDP-ribitol. Interestingly, a group in the U.S. reported that oral administration of ribitol to muscular dystrophy model mice with FKRP point mutations restored dystroglycan glycosylation, resulting in a therapeutic effect10. The reason may be that the enzymatic reaction is promoted when the amount of substrate (CDP-ribitol) is increased even in mice with mutant FKRP, but the enzymatic kinetics of the reaction has not been demonstrated. Following positive results in mouse experiments, clinical trials for limb-girdle muscular dystrophy 2I, in which FKRP is the causative gene, have already been initiated in the U.S. Although the efficacy of this ribitol therapy may depend on the type of causative gene mutation, it is undoubtedly a revolutionary advance in therapy for dystroglycanopathy for which no fundamental treatment exists.
It has indeed taken 15 years since the discovery of dystroglycanopathy to fully elucidate the glycan structures and modifying enzymes. Currently, the efficacy of several therapeutic strategies proposed based on the elucidated pathomechanism has been verified, and expectations for future clinical application are high. In addition, although not introduced in this article, studies using several different mouse models of dystroglycanopathy have revealed the physiological and pathological significance of ribitol phosphate modification11. The biological importance of ribitol phosphate modification is expanding, leading to high expectations for future developments.



