
Takaharu Mori
Senior Research Scientist at the Theoretical Molecular Science Laboratory, RIKEN Cluster for Pioneering Research
In 2008, Dr. Mori completed his doctoral course and obtained a PhD in science from the Department of Physics, Graduate School of Science, Nagoya University. He worked as a RIKEN special postdoctoral researcher and assumed his current position in 2019. His main research interests include developing novel algorithms for molecular simulations and protein structure modeling from cryo-electron microscopy data.

Yuji Sugita
Chief Scientist at the Theoretical Molecular Science Laboratory, RIKEN Cluster for Pioneering Research
In 1998, Dr. Sugita completed his doctoral course and obtained a PhD in science from the Department of Chemistry, Graduate School of Science, Kyoto University. He served as an assistant at the Institute for Molecular Science and a lecturer at the Institute of Molecular and Cellular Biosciences, University of Tokyo, and assumed his current position since 2012. His research interests include developing new methodologies for theoretical molecular science and exploring biological phenomena in the cellular environment using computational science.
The recent COVID-19 pandemic is caused by a new coronavirus, SARS-CoV-2, whose spike protein on its surface binds to human cells via angiotensin-converting enzyme 2 (ACE2) receptors in the initial stage of infection. In that process, the structure of the spike protein changes from a “down-form” to an “up-form”, as demonstrated by cryo-electron microscopy. Biochemical experiments have shown that the surface of the spike protein is modified by many glycans. Glycans have been considered to play a role in protection against antibodies, i.e., immune evasion; however, how they contribute to the structural changes of the spike protein remains unclear. In this paper, we describe the role of glycans as elucidated by molecular dynamics simulation of the spike protein.
The spike protein is a large membrane protein protruding from the surface of coronavirus in an arrangement that resembles a crown (corona in Greek), hence the name, coronavirus. The spike protein occurs as a homo-trimer generally comprising two subunits S1 and S2 (Figure 1A). S1 consists mainly of an N-terminal domain (NTD) and receptor binding domain (RBD), the latter binding to ACE2 receptors in host cells. ACE2 receptors are abundantly expressed in human organs such as the heart, lung, and kidney, and oral mucosal membranes such as the membrane on the surface of the tongue. Although the ACE2 enzyme plays a role in blood pressure control, it also binds to the spike protein, providing a route for coronavirus entry into cells1
To date, the conformations of diverse forms of the spike protein have been clarified by X-ray crystallography and single-particle cryo-electron microscopy (cryo-EM)2-5. The two major structural forms of the spike protein are the “down-form” and “up-form”. There has been increasing evidence showing that binding of the spike protein to ACE2 receptors stabilizes the up-form (Figure 1B)6. Furthermore, biochemical experiments have shown that many asparagine residues on the surface of the spike protein are modified by glycans7. Because wide coverage of the spike protein surface by glycans makes antibody attacks unlikely, glycans are considered to play a role in immune evasion by not only the new coronavirus, but also various other viruses such as influenza virus and HIV8. Because glycans are highly flexible, on the other hand, it is difficult to analyze the detailed conformation of individual glycan molecules even with the use of cryo-EM. To date, there has been poor understanding of the effect of modifications by glycans on the structural change of spike protein. To elucidate the molecular mechanisms underlying the structural change of spike protein, the authors performed molecular dynamics (MD) simulations of the spike protein. An MD simulation refers to a method of predicting molecular structures and motions by building a virtual molecular system in the computer, and applying Newton’s equation of motion F=ma to individual atoms. The force F is calculated from the first derivative of the potential energy with respect to the atomic position. Potential energy of the system, also known as the force field, is generally calculated using the following equation: Terms 1 to 5 represent covalent bond stretching motion and deformation motion, dihedral angle rotation energy, van der Waals interaction, and Coulomb interaction, respectively. In an MD simulation using the all-atom model, one atom is represented by one sphere, and the system is time-evolved with one step set at approximately 2 femtoseconds to accurately integrate the bond stretching motion. Many biological phenomena, on the other hand, occur on a time scale of microseconds to milliseconds or more; therefore, time integration for several hundred million steps or more is required to observe biologically important phenomena by MD simulations. Therefore, when a large protein is the study subject, high performance MD software and powerful computers, such as a supercomputer, are required. Careful modeling of the target system is important for reliable MD simulations. Ideally, the simulation system should mimic the biological environment or experimental conditions as much as possible. In the case of glycoproteins the amino acids should be modified with glycans, and in the case of membrane proteins the protein should be embedded in lipid bilayers. CHARMM-GUI (https://www.charmm-gui.org)9 is in wide use as a tool for modeling various complex systems. For example, one function in CHARMM-GUI is designed to build a conformation of glycan by visually joining various monosaccharides10. It is also capable of automatically generating input files of major MD software thus allowing not only theoreticians, but also experimentalists to easily prepare for MD calculations for glycans and glycoproteins. The molecular system used in this study is shown in Figure 2. Although the spike protein is essentially a membrane protein, we simulated a water-soluble moiety, which was immersed in 150 mM NaCl solution in this study. Two systems (down-form, up-form) were provided, and based on experimental data, amino acids at 66 sites were modified with glycans. Each entire system comprises approximately 760,000 atoms, including water molecules, representing calculations on a larger scale than for recent typical systems comprised of 200,000 to 300,000 atoms. In this study, we used the MD software GENESIS (https://www.r-ccs.riken.jp/labs/cbrt/), which has been mainly developed by the RIKEN Computational Biophysics Research Team. GENESIS is capable of dealing with a wide range of biomolecules, including proteins, lipids, nucleic acids, and glycans, for molecular to cellular scale simulations11-12. In addition to the conventional MD algorithms, a wide variety of functions are available, including the generalized-ensemble algorithms, cryo-EM flexible fitting, quantum mechanics / molecular mechanics (QM/MM) methods, and free energy calculation methods aiming at drug discovery13-16. Recently, Drs. Jaewoon Jung and Chigusa Kobayashi worked to optimize the performance of GENESIS on the Fugaku supercomputer and succeeded in improving it by more than 100 times that on the K computer. In this study, we performed MD simulations for the down-form and up-form structures in a microsecond each using GENESIS, Fugaku, and Oakforest-PACS at the University of Tokyo. Based on the trajectory data obtained from the MD simulations, amino acid - amino acid interactions and amino acid - glycan interactions were analyzed comprehensively. As a result, glycans that modify three aspartic acids N165, N234, and N343 were found to be contributory to the structural stabilization of the down-form and up-form. Figure 3 shows the major inter-residue interaction pairs between RBD, NTD, and S2 domains. The N343 glycan (Figure 3A, green) was found to form hydrogen bonds with the adjacent RBD to join RBDs together in the down-form, and the interaction is lost upon conversion to the up-form. The N165 glycan (Figure 3B, orange) interacted with an RBD in close contact with the NTD, covering the RBD from above; even when the RBD converted from the down-form to the up-form, the N165 glycan came into contact with the RBD while flexibly changing its structure. We found that when the down-form converts to the up-form, the RBD-S2 interaction is broken completely (Figure 3C), the N234 glycan (Figure 3B, yellow) goes under the RBD and comes into contact with S2. It can be conjectured that as the N234 glycan intrudes between the RBD and S2 like a “tension pole”, return from the up-form to the down-form is made unlikely. In many cases, electrostatic interaction is a driving force of the structural change of proteins. We analyze the electrostatic potential of the down-form obtained from the MD simulations, using the adaptive Poisson-Boltzmann solver (APBS)18. The results showed that the RBD-RBD interface is positively charged over a broad range due to presence of the arginine and lysine residues (Figure 4). Hence, it can be conjectured that the repulsive interactions between those positively charged residues had accumulated as a frustration at the RBD-RBD interface of the down-form. This seems to have served as a driving force for the RBD structural changes as follows: The N343 glycan that joins the RBDs is detached by thermal fluctuation to promote structural change to the up-form (Figure 3A), and the N234 glycan intrudes under the RBD to stabilize the up-form (Figure 3B). Combining the above findings, we proposed the molecular mechanisms underlying the structural change of the spike protein illustrated in Figure 517. This study suggested that glycans play a key role in structural changes of the spike protein. MD calculations for proteins involved in COVID-19 are being made worldwide, and the focus of recent vigorous research activities has been on the analysis of the spike protein variants19-20. There has also been increasing understanding of the roles of glycans in complexes with ACE2 receptors; for example, two types of ACE2 receptor glycans have been shown to occur: one that promotes binding to RBD, and the other that inhibits the same21. We hope compilation of findings based on such molecular structures will lead to a new drug design strategy that takes into consideration glycan structures and dynamics. This study was conducted jointly with Drs. Jaewoon Jung and Chigusa Kobayashi at the Computational Biophysics Research Team, RIKEN Center for Computational Science; Dr. Hisham Dokainish at the Theoretical Molecular Science Laboratory, RIKEN Cluster for Pioneering Research; and Dr. Suyong Re at the Laboratory for Biomolecular Function Simulations, RIKEN Center for Biosystems Dynamics Research (currently at the National Institutes of Biomedical Innovation, Health and Nutrition).
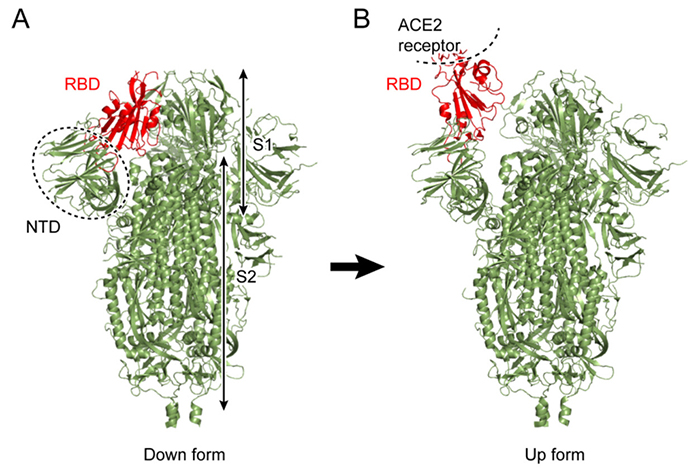
Figure 1. Conformational forms of the new coronavirus spike protein(A) Down-form, (B) up-form. The receptor-binding domain (RBD) is shown in red.
2. Molecular dynamics simulations of the spike protein
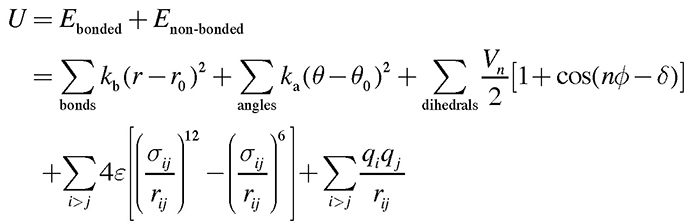
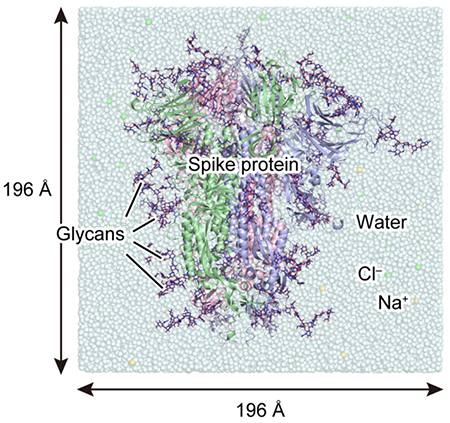
Figure 2. System used for MD simulationsHere, the three polypeptide chains A, B, and C of the spike protein are represented by different ribbon models in red, green, and blue, respectively; and a glycan, by a navy-blue stick model. The box size of each dimension is approximately 196 Å .
3. Analysis of amino acid - amino acid and amino acid - glycan interactions
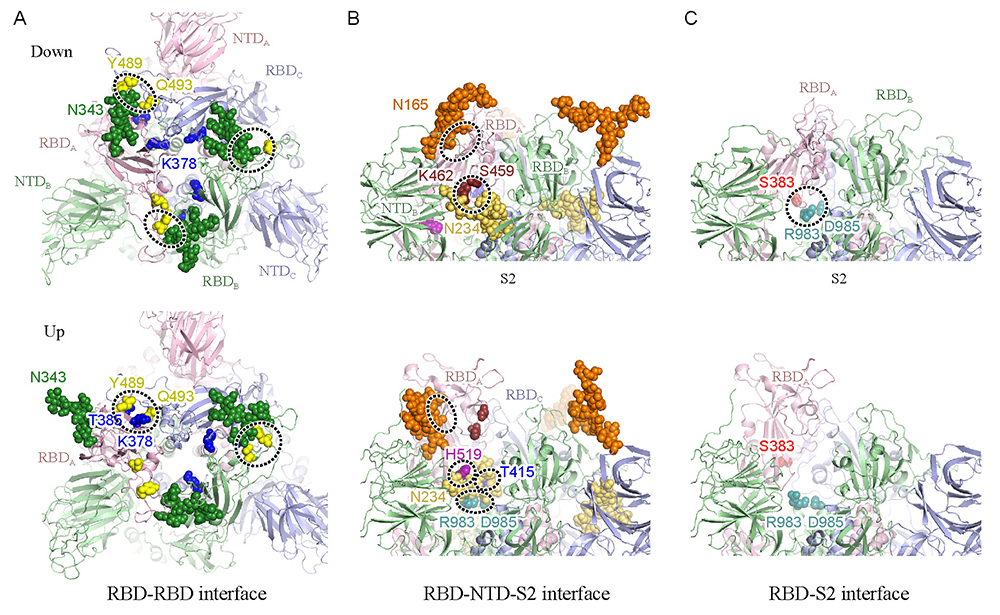
Figure 3. Snapshots of simulated structural changes in 1 microsecond(A) The spike protein viewed from above, with the RBD-RBD interface remarked; (B) the upper portion of the spike protein viewed from one side, with the RBD-NTD-S2 interface remarked; (C) the RBD-S2 interface remarked. The upper panel shows the down-form, and the lower panel shows the up-form. Important glycans are shown by sphere models, with sites of relatively strong interactions enclosed by dotted borders. Cited from Reference 17.
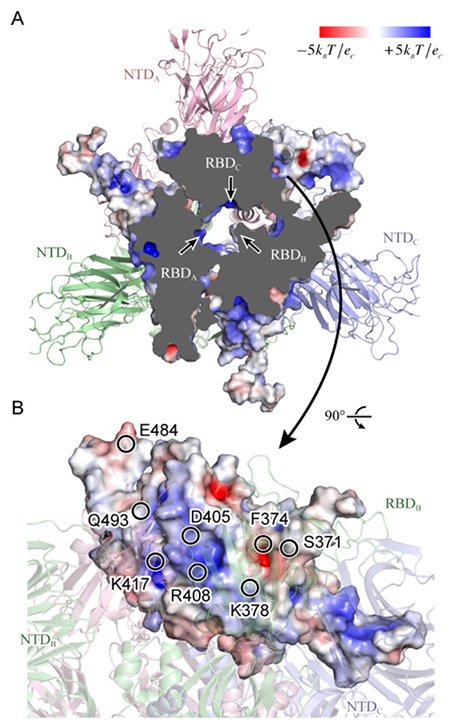
Figure 4. Static potential of spike protein RBD(A) Looking from above the spike protein, the upper portion has been cut away to increase inside visibility. The three arrows each indicate a positively charged moiety at the RBD interface. (B) Looking from the side of RBDC, the representative amino acids in the vicinity of a positively or negatively charged moiety are shown by circles. Cited from Reference 17.
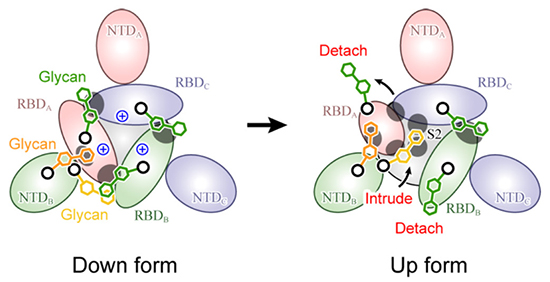
Figure 5. Molecular mechanism for spike protein structural changesThis is a schematic diagram of the spike protein viewed from above, with the glycans contributing to structural stabilization shown by green (N343), orange (N165), and yellow (N234) hexagons. The gray circles indicate the places of strong protein-glycan interaction.
4. Future Prospects
Acknowledgments
References



