
Kenji Kadomatsu
The author graduated from Kyushu University School of Medicine in 1982. 2004-present, Professor at Nagoya University Graduate School of Medicine (Department of Biochemistry). 2017-present, Dean of Nagoya University School of Medicine and Graduate School of Medicine. As of 2020, President of the Japanese Society of Carbohydrate Research, and Director of the Japanese Biochemical Society. The author discovered the growth factor midkine, which has led to his interest in two areas of research: glyco-neuroscience and pediatric neuroblastoma.
Neurons are cells that are fully differentiated and do not divide further. Therefore, it is difficult to grow these cells, once damaged, and to replace them with new ones. Neurons with injured axons can only maintain their function by regenerating their axons. However, little or no axon regeneration takes place in the central nervous system. It has been revealed that glycans, glycan receptors, and autophagy are involved in this regeneration process. In this article, I describe the timeline and historical development of this research topic in a way that makes it more readable than the typical review article in an academic journal.
One hundred years ago, the Spanish Nobel laureate and neuroanatomist Santiago Ramon y Cajal sketched the distorted spherical shape of the axon terminal of a damaged central nerve. This structure is called a “dystrophic endball” (there are other names such as retraction bulb, frustrated growth cone, or dystrophic growth cone). Cajal indicated that “once injured, axons do not regenerate”, and also, “if this issue could be solved, it would be due to the great efforts of scientists in future generations”. Even now, the axon regeneration problem has yet to be sufficiently solved.
If axonal damage occurs, the axon on the peripheral side of the injury site would be rendered useless by Wallerian degeneration, while the axon on the cell body side would survive for decades (Ruschel, 2015). A dystrophic endball forms at each axon’s terminal, but this structure never extends (Figure 1). Why doesn’t the axon regenerate? There are two main reasons for this. One is that central neurons have low axonal regenerative capacity. Regenerative capacity of these cells has been vigorously studied, and the mechanism has been partly elucidated. However, there is too much information on this topic to mention here. I refer readers to other review articles (Kadomatsu, 2014; Sakamoto, 2017). The other is the emergence of regeneration-inhibiting molecules. These molecules can be classified into the following three main categories.
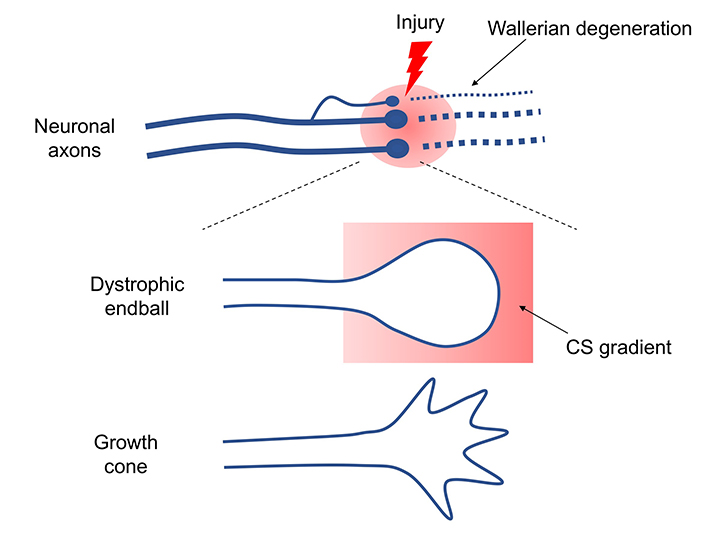
Does the dystrophic endball, the structure found by Cajal, really form? If a dye is injected into a rat's cortical motor cortex, it is taken up by primary motor neurons and transported to the axons in about a week. When my colleague and I injected spinal cord-injured rats and observed the dye’s uptake at the injury site, surprisingly, nearly all of the axon terminals of injured motor nerves formed dystrophic endballs (Sakamoto, 2019). This result definitely demonstrated that Cajal had beautifully captured the phenomenon in-vivo. Culturing adult dorsal root ganglion (DRG) neurons in culture dishes containing CS distributed in a concentration gradient can reproduce dystrophic endball formation in vitro (Tom, 2004). Adult DRG neurons attempt to extend their axons against the CS gradient, but the axons stop extending at some point, form dystrophic endballs, and retract in the opposite direction (Sakamoto, 2019). By contrast, the tips of axons of adult DRG neurons cultured using laminin as a matrix adopt an outstretched hand-like morphology (growth cone) and extend without stopping.
Then, by comparing the growth cone to the dystrophic endball, we noticed that the latter has many vesicles. These vesicles turned out to be LC3-II positive autophagosomes. We further examined the reasons for this autophagosome accumulation and found that the fusion of autophagosomes and lysosomes was inhibited and autolysosomes were not formed; in other words, autophagy was interrupted (Sakamoto, 2019). Inhibition of autophagosome/lysosome fusion using inhibitory agents or by knockdown of snare proteins led to dystrophic endball formation, indicating that this interruption of autophagy is also a sufficient condition for dystrophic endball formation.
Several molecules have been reported to act as CS receptors, among which PTPRσ and LAR (receptor-type tyrosine phosphatases) are important (Shen, 2009; Coles, 2011). If the CS → PTPRσ axis causes dystrophic endball formation, then the molecules required for dystrophic endball formation would be expected to be substrates for PTPRσ. We therefore turned our attention to cortactin. This is because cortactin undergoes tyrosine phosphorylation, which is required for actin polymerization, and this polymerization is essential for the fusion of autophagosomes and lysosomes (Hasegawa, 2016). Indeed, phosphorylated cortactin was shown to be a substrate of PTPRσ, and in addition, dephosphorylation of cortactin was markedly enhanced in dystrophic endballs when compared with growth cones. Furthermore, knockdown of cortactin induced dystrophic endball formation. These findings are the basis for the following mechanism: CS → PTPRσ → dephosphorylation of cortactin → interruption of autophagy → dystrophic endball formation → inhibition of axon regeneration (Sakamoto, 2019) (Figure 2).
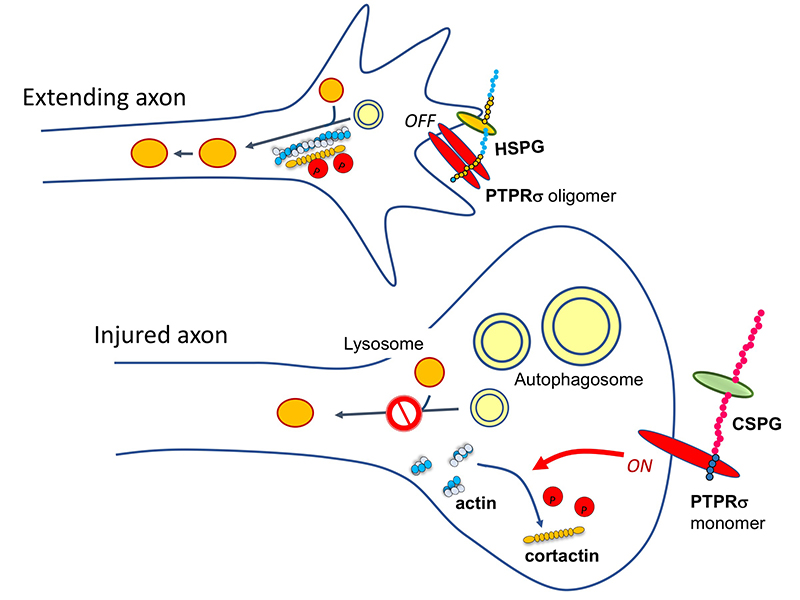
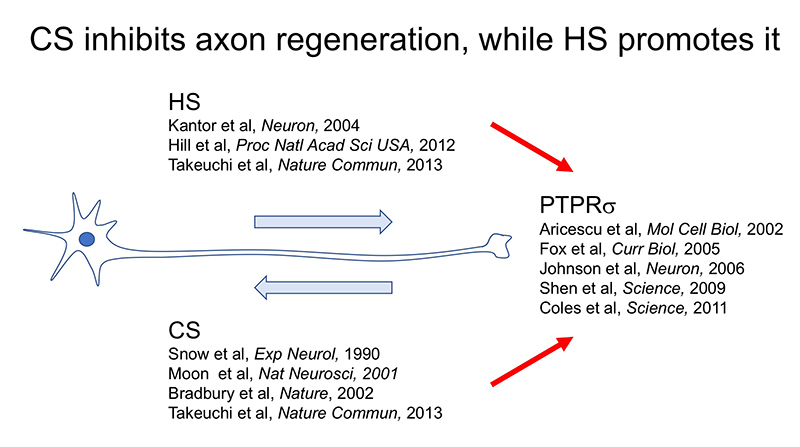
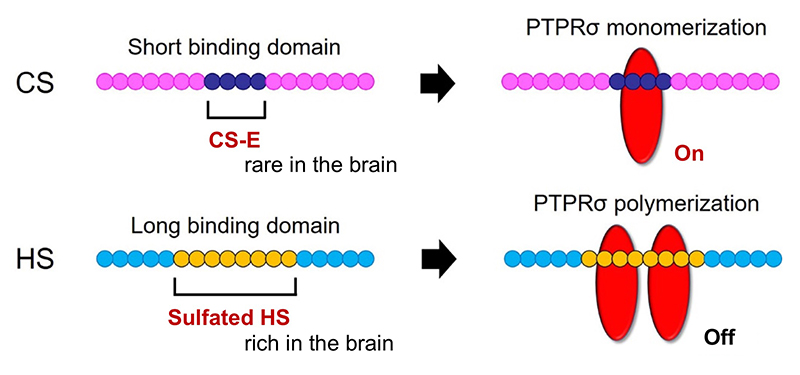
Here’s another mystery: the CS-HS paradox. PTPRσ and LAR bind not only to CS, but also to heparan sulfate (HS) (Aricescu, 2002; Fox, 2005; Johnson, 2006; Shen, 2009). Moreover, CS and HS bind to the same site on these receptors (Shen, 2006; Coles, 2011). Nevertheless, CS inhibits axon regeneration (Snow, 1990; Moon, 2001; Bradbury, 2002), whereas HS promotes regeneration (Kantor, 2004; Hill, 2012) (Figure 3). Consistent with these findings, knockout, in mice, of the enzyme chondroitin sulfate N-acetylgalactosaminyltransferase-1, which is at the bifurcation point for CS and HS biosynthesis, leads to elimination of CS, increase in HS, and greater enhancement of axon regeneration and restoration of nerve function than CS degradation treatment alone (Takeuchi, 2013). One of the clues to solution of this CS-HS paradox is the hypothetical action of PTPRσ as a molecular switch (Coles, 2011). Presumably, HS induces oligomerization of PTPRσ and thereby turns off its enzymatic activity, while CS turns it on by mediating its monomerization. However, it remains unclear why and how such regulation occurs. Natural CS and HS have various sulfation patterns and are considered to have their own specific functions. In CS, sulfuric acid is added to disaccharide repeats of galactose and N-acetylgalactosamine and the CS structure known as the E-unit (CS-E) is sulfated in both the C4 and C6 positions of N-acetylgalactosamine. In the cerebrum, this structure is rare, with an abundance of only about 3%. If the total length of a CS chain is assumed to be 100 disaccharide units, then the frequency of a tetrasaccharide consisting of tandem CS-Es is expected to be about one per chain. The CS-E was the only CS unit to show a high affinity for PTPRσ. Moreover, tetrasaccharidal CS-E caused PTPRσ to monomerize and turned on its phosphatase activity (Figure 4). On the other hand, HS does not bind to PTPRσ in the absence of sulfation, but most of the sulfation patterns show affinity. As approximately 50% of the HSs are sulfated in the cerebrum, the length of HS bound to PTPRσ is expected to be sufficiently long. It was also found that the sulfated tetrasaccharidal HS had lower affinity for PTPRσ than tetrasaccharidal CS-E, and this affinity was similar to that of octasaccharides. Octasaccharidal HS actually caused PTPRσ to multimerize and turned off its enzymatic activity (Figure 4). Overall, in the early stages, HSPG on the neuronal axon membrane causes PTPRσ to multimerize, allowing autophagy to proceed without problem. However, when an injury occurs and CSPG appears, a short stretch in the CS chain (the main component seems to be CS-E) induces PTPRσ to monomerize, triggering the following cascade: dephosphorylation of cortactin, failure in actin polymerization, inhibition of autophagosome-lysosome fusion, dystrophic endball formation, and inhibition of axon regeneration (Figure 2).
Knowing the mechanisms that inhibit axon regeneration will help to overcome the challenges posed by the inhibition, and will lead to practical, never before dreamed of, treatments of neuronal injuries. A number of interventions such as CS-degrading enzymes and PTPRσ inhibitors are being tested in practice (Lang, 2015; Warren, 2018). Notably, Silver's group reported that a single dose of a CS-degrading enzyme, chondroitinase ABC, could improve paralysis in the chronic phase of high-level spinal cord injuries associated with diaphragmatic paralysis (Warren, 2018). This report is the culmination of CS-related research efforts, and is of great significance for expanding the potential of treatment for chronic spinal cord injury.
Meanwhile, the findings from the study of CS-PTPRσ are also important from a physiological perspective. For example, the secretion of proteolytic enzymes is essential for extracellular matrix degradation required for axon regeneration, and this secretion is dependent on cortactin (Clark, 2007). Cortactin-dependent secretion is consistent with the fact that PTPRσ-dependent inactivation of cortactin via dephosphorylation impedes axon regeneration. In addition, Tran et al. provided an interesting discussion in their review (Tran, 2020): Axons do not grow indefinitely, but the growth must be stopped as the axons approach the dendrites of target neurons to form synapses. As CSPG is also secreted from neurons, the CS-PTPRσ axis may be operational in the dendrite arena. Further, PTPRσ in the axon tip binds to molecules such as TrkC, NGL-3, and NT-3 existing on the surface of dendrites (Coles, 2014; Naito, 2017) and is likely to play an important role in synapse formation. In other words, the CS-PTPRσ axis should not be viewed solely from a pathological viewpoint (inhibition of axon regeneration), since this regulatory axis may also have a physiological function (Tran, 2020).
The inhibitory mechanism of axon regeneration, involving CS and PTPRσ, is important from another perspective: the axon tip-autophagy relationship. In normal neurons, autophagy begins at the tip of the axon, and autophagosomes and autolysosomes are transported back to the cell body to complete autophagic flow (Wong, 2015). Thus, the discovery that interruption of this flow at the axon tip caused dystrophic endball formation and inhibition of axon regeneration once again highlighted the importance of autophagy in neurons. This important insight applies to neurodegenerative diseases as well. For instance, mutations in PINK1 and Parkin genes encoding respectively kinase and E3 ubiquitin ligase have been found in familial Parkinson's disease, and these proteins are essential in mitochondrial autophagy (mitophagy) (Clark, 2006; Park, 2006; Yang, 2006). There is also a generally accepted concept that synaptic degeneration precedes neuronal cell death in neurodegenerative diseases. Autophagosomes accumulate during the synaptic degeneration accompanying neurodegeneration. The extent to which these phenomena and the CS-PTPRσ axis overlap is another interesting question.



