Katsuhiko Yanagisawa , M.D.
Research Institute, National Center for Geriatrics and Gerontology
One of the fundamental questions regarding the pathogenesis of Alzheimer disease (AD) is how soluble amyloid β-protein (Aβ), a major component of senile plaques, starts to assemble into toxic amyloid fibrils in the brain. Accumulating evidence suggests that Aβ binds to ganglioside on neuronal membranes, leading to formation of an endogenous seed with an altered conformation (ganglioside-bound Aβ; GAβ) for amyloid fibril formation in the brain 1,2. Notably, the risk factors for the development of AD, including aging and expression of apolipoprotein E4, likely facilitate the formation of ganglioside-rich, lipid raft-like membrane microdomains at presynaptic terminals, which provide a favorable milieu for the GAβ generation 2. Furthermore, it has been suggested that endocytic pathway abnormality of neurons, which was previously observed in the neurons of AD brains, is involved in the formation of the ganglioside-rich membrane microdomains 2. Recently, the conformational change of Aβ following interacting with GM1 has been validated by ultra-high-field NMR analyses 3. Additionally, it has also been reported that amyloid fibrils formed on GM1-containing, lipid raft-like membrane microdomains, are more toxic to neuron-like cells (PC12) than those formed in solutions 3. These lines of evidence may provide new insights into the molecular pathogenesis shared by various human amyloidoses, including AD.
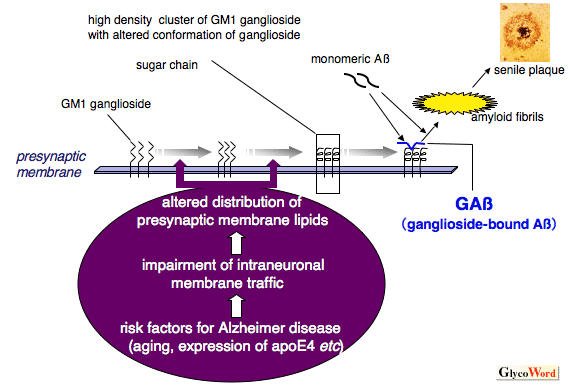
Figure Putative mechanism underlying ganglioside-induced amyloid fibril formation in Alzheimer



