
Tadanobu Takahashi
Department of Biochemistry, School of Pharmaceutical Sciences, University of Shizuoka
Tadanobu Takahashi graduated from the School of Pharmaceutical Sciences, University of Shizuoka, in 1999. He completed his master痴 degree at the Graduate School of Pharmaceutical Sciences, University of Shizuoka, in 2001, and obtained his Ph.D. in 2004 from the Graduate School of Pharmaceutical Sciences, University of Shizuoka. He worked as a research fellow in the Core Research for Evolution Science and Technology (CREST) program in Japan Science and Technology Agency from 2004 to 2006, was assistant professor in the Department of Clinical Molecular Cell Biology from 2006 to 2007, and is currently at the Department of Biochemistry, School of Pharmaceutical Sciences, University of Shizuoka.

Takashi Suzuki
Department of Biochemistry, School of Pharmaceutical Sciences, University of Shizuoka
Takashi Suzuki graduated from the School of Pharmaceutical Sciences, University of Shizuoka, in 1979. He completed his master痴 degree at the Graduate School of Pharmaceutical Sciences, University of Shizuoka, in 1981, and obtained his Ph.D. in 1984 from the Graduate School of Pharmaceutical Sciences, University of Shizuoka. He worked as a research fellow at the Shino Test Institute from 1984 to 1990. He moved to the Department of Biochemistry, School of Pharmaceutical Sciences, University of Shizuoka, as assistant professor in 1991, appointed lecturer in 1994, associate professor in 1996, and from 2007 to the present has been professor at the same university. In 1998-1999, he worked at St. Jude Children痴 Research Hospital, U.S.A., as a visiting scientist and a research worker abroad supported by the Ministry of Education, Science, and Culture of Japan
A sulfated glycolipid, 3-O-sulfogalactosylceramide (sulfatide), is ubiquitously present in various tissues such as the brain, kidney, respiratory tract and gastrointestinal tract. Sulfatide is localized mainly in the Golgi apparatus, cellular membrane, and lysosomes, or cytosol especially in many cancer cell lines. The metabolism of sulfatide is very simple compared to that of many gangliosides. The synthesis of sulfatide starts at the endoplasmic reticulum (ER) by the addition of galactose from UDP-galactose to ceramides. This reaction is catalyzed by the UDP-galactose: ceramide galactosyltransferase (CGT; EC 2. 4. 1. 45)1. Galactosylceramide (GalCer), which is the precursor of sulfatide, is transported to the Golgi apparatus, where sulfatide is finally synthesized by 3’-phosphoadenosine-5’-phosphosulfate (PAPS): cerebroside sulfotransferase (CST; EC 2. 8. 2. 11)2. CST has been reported to be a homodimeric protein localized to late Golgi apparatus3. The sulfate group of sulfatide is hydrolyzed by arylsulfatase A (ASA; EC 3. 1. 6. 8) in lysosomes. This reaction requires the help of a sphingolipid activator protein-1 (SAP-1), also called saposin B (SapB), which extracts sulfatide from membranes and thereby making it accessible to ASA4 (Fig. 1). Recently, an alternative sulfatase-independent pathway of sulfatide degradation has been suggested by indications in a neuroblastoma cell line for direct generation of ceramide from endocytosed sulfatide without prior desulfation5. Sulfatide exhibits various structures, including different lengths of the acyl chain and ceramide moiety, which can also be hydroxylated as well as other sphingolipids.
Deficiency of the lysosomal enzyme ASA6 or mutations in the gene coding for SapB7 cause metachromatic leukodystrophy (MLD), a demyelinating disease, due to the accumulation of lysosomal sulfatide. Sulfatide is a biologically multifunctional glycolipid involved in the nervous system, diabetes, the immune system, hemostasis/thrombosis, bacterial infection and virus infection such as human immunodeficiency virus. We found that sulfatide promotes translocation of newly synthesized viral ribonucleoprotein (vRNP) complexes with viral nucleoprotein (NP) of influenza A virus from the nucleus to cytoplasm and progeny virus production by association with a viral surface glycoprotein, hemagglutinin (HA), delivered to the cell surface8. In this review, we describe the role of sulfatide in influenza A virus infection.
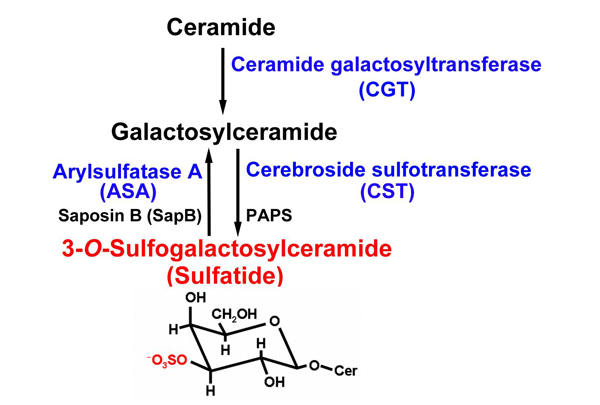
Fig. 1 Metabolism of sulfatide.
The chemical structure of sulfatide is shown. Sulfatide (red characters) is synthesized from cerimide through galactosylceramide by two transferases (CGT and SCT). Then the sulfate group of sulfatide (red atoms in the sulfatide structure) is degraded by ASA and metabolized to galactosylceramide. All enzymes essential for sulfatide metabosim used in our study are shown in blue.
Influenza A virus infects and is transmitted among avian and mammalian animals including human. Influenza A virus recognizes and binds to various sialo-glycoconjugates on the host cell surface via the viral envelope glycoprotein, HA. Sialic acid plays an essential role as the virus receptor in the influenza A virus life cycle. Another envelope glycoprotein, neuraminidase (NA), has sialidase activity for cleaving sialic acid residue and facilitates release of the progeny viruses from the cell surface membrane9. Recently, it has been suggested that sialidase activity of NA under viral entry into cells through endocytosis plays an important role in the virus infection and replication10, 11. Sulfatide has been ubiquitously detected in various epithelial cell lines and animal tissues that permit sufficient influenza A virus replication. We have found that influenza A virus binds to sulfatide, which has no sialic acid12.
We investigated the function of sulfatide in the influenza A virus infection cycle by knockdown of sulfatide expression in Madin-Darby canine kidney (MDCK) cells, which are known to adequately support the virus replication, and by genetic up-regulation of sulfatide expression in COS7 cells, which lack sulfatide expression and sufficient influenza A virus replication.
Specifically, we genetically produced sulfatide-enriched cells from parental COS7 cells defective in sulfatide. Sulfatide accumulation leads to a feedback inhibition of GalCer synthesis. We therefore transfected COS7 cells with both CST and CGT genes from MDCK cells and established two cell clones (SulCOS1 and SulCOS2) that stably express sulfatide. Enhancement of influenza A virus (A/WSN/33 H1N1 strain) multiple replications in sulfatide-enriched cells was clearly confirmed by immunostaining of the cells (Fig. 2). Progeny virus titers of the supernatant from cell clones were increased by 500-3000 times in comparison with that from the parent COS7 cells, which showed little replication of the virus. In contrast, sulfatide-enriched cells showed reduction of about 28% in the initial infection in comparison with that of the parent cells.
To evaluate the impact of sulfatide depletion on influenza A virus replication, we generated sulfatide-knockdown cells from MDCK cells by transfection with the ASA gene and by RNA interference (RNAi) targeting CST mRNA. We obtained MDCK cells stably expressing the ASA gene. The sulfatide-knockdown cells showed an approximately 60-fold reduction in progeny virus titers generated by the virus replication and a slight increase in initial infection. Application of RNAi targeting endogenous CST mRNA to MDCK cells also resulted in a significant reduction of the virus replication. Sulfatide-enriched cell clones also showed increased replication of other influenza A virus strains regardless of viral subtype (H1N1, H3N2 or H5N3) or host (human or avian). Taken together, these results indicate that sulfatide expression in mammalian cells greatly enhances multiple replications of influenza A virus and that sulfatide is not the initial receptor in the virus infection.
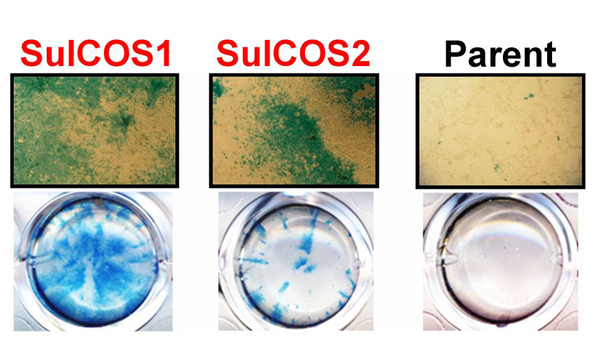
Fig. 2 Sulfatide promotes influenza A virus replication.
Two sulfatide-enriched cell lines (SulCOS1 and SulCOS2; red characters) were generated by transfection to the parent cells, which could not synthesize sulfatide, with two sulfatide-synthesis transferase (CGT and CST) genes. Influenza A virus-infected cells are observed by blue staining of viral antigens. Cells cultured for 24 h at 37ºC after infection are shown in images under optical microscopy at a magnification of 40 (upper) and in images of wells in culture plates (lower). Virus replication in the sulfatide-enriched cells is much higher than in the parent cells.
We previously established a mouse anti-sulfatide monoclonal antibody (named GS-5)13. To determine whether GS-5 has an inhibitory effect on influenza A virus infection, MDCK cells were treated with GS-5 before, at the same time as, and after virus infection. Treatment with GS-5 after virus infection induced a great reduction in virus multiple replications, but GS-5 treatment before and at the same time as virus infection had no effect on viral initial infection and replication. Additionally, GS-5 treatment induced distinct inhibition of virus replication for 4-24 h but not for 0-2 h after virus infection. GS-5 also had the same effect on other strains independent of viral subtype (H1N1, H3N2 or H5N3) and host (human or avian). These results suggest that sulfatide on the cell surface is involved in influenza A virus replication.
To assess whether the anti-sulfatide antibody can inhibit virus replication in vivo, C57BL/6 mice were intranasally infected with pathogenic influenza A virus A/WSN/33 (H1N1) strain. GS-5 was administered intranasally once daily for 5 days beginning 1 day preinfection. Administration of GS-5 protected 50% of mice against 100% lethal challenge with pathogenic influenza A virus and markedly reduced progeny virus titers in the lungs of the mice.
NP of influenza A virus forms vRNP complexes together with the viral polymerases (PB1, PB2, and PA), and viral genome RNA segments in the nucleus and the vRNP complexes are then exported to the cytoplasm. On the other hand, two surface spike glycoproteins, HA and NA, are transported to the cellular surface membrane through an exocytic pathway. In sulfatide-enriched cells (SulCOS1) at 7 h after infection at 37ºC, most of the newly synthesized HA and NA was localized to the cell surface, and most of the newly synthesized viral NP had translocated from the nucleus to cytoplasm. In the parent cells, most of the newly synthesized viral HA and NA was localized to the cell surface, but most of the newly synthesized viral NP remained in the nucleus. In sulfatide-enriched cells that were treated with GS-5 after virus infection, most of the newly synthesized viral NP remained in the nucleus despite normal localization of newly synthesized viral HA. The results suggest that sulfatide, which is expressed on the cell surface during the process of influenza A virus replication, promotes nuclear export of vRNP complexes, resulting in a significant efficient formation of infectious progeny virus.
To elucidate the involvement of HA and NA in nuclear export of the viral NP regulated by sulfatide, SulCOS1 cells infected with influenza A virus A/Memphis/1/71 (H3N2) strain were treated with anti-H3 HA MAb (2E10), which blocks the binding between the virus and sulfatide. Translocation of the newly synthesized NP from the nucleus to cytoplasm in the cells was almost completely suppressed by 2E10, similar to the effect of GS-5. In contrast, anti-H3 HA MAb (1F8), which does not block the binding of IAV to sulfatide, and anti-N2 NA MAb (SI-4) as well as TU-1 did not suppress nuclear export of the viral NP. These results indicate that association of sulfatide with HA delivered to the cell surface induces translocation of the newly synthesized viral NP from the nucleus to cytoplasm.
To clarify the binding activity of HA to sulfatide, we selected a baculovirus protein expression system to obtain a large amount of HA glycoprotein in influenza A virus A/duck/Hong Kong/313/4/78 (H5N3) strain. The recombinant HA expressed in insect cells was extracted by detergent and purified by affinity column for the histidine tag sequence added to the C-terminus of the HA. The recombinant HA expressed on insect cells showed hemadsorption activity of red blood cells and reaction with specific monoclonal antibody. The purified recombinant HA exhibited high hemagglutination unit itself and hemagglutination inhibition after reaction with specific monoclonal antibody. These results indicate that the recombinant HA expressed in insect cells possesses both receptor binding ability and antigenicity similar to influenza A virus. The recombinant HA bound to sulfatide in a dose-dependent manner as well as α2,3-sialylparagloboside, which was preferentially recognized by avian influenza A virus. The binding of the recombinant H5 HA to sulfatide was prevented by re-incubation with anti-H5 HA MAb, but not with anti-H3 HA MAb, in a dose-dependent manner. The anti-H5 HA MAb, which probably recognizes the epitope neighboring the receptor binding site on each HA and prevents viral hemagglutination by steric hindrance of HA binding to the sialic acid moiety, inhibited binding of the recombinant H5 HA to sulfatide. The results indicate that HA binds to sulfatide, possibly through the region near the receptor binding site on HA14. On the other hand, sulfatide also inhibits sialidase activity of viral NA under acidic condition only15. It may be necessary to investigate the interaction of not only HA but also NA with sulfatide.
Our data show that sulfatide promotes translocation of the newly synthesized vRNP complexes from the nucleus to cytoplasm by association with HA delivered to the cell surface8 (Fig. 3). Administration of anti-sulfatide antibody resulted in strong inhibition of influenza A virus replication, even after virus infection, and protected mice against lethal challenge with the virus. The development of antiviral drugs will probably play a critical role in the next influenza pandemic. NA inhibitors such as oseltamivir (Tamiflu) are currently being used for protection against and treatment of influenza. However, an oseltamivir-resistant strain (a Russian H1N1 type) that emerged in North Europe in 2007 spread worldwide until 2008. The emergence of such drug-resistant viruses has become a serious problem. Most current human influenza A viruses have also become resistant to viral M2 ion channel inhibitors such as amantadine. The binding inhibitors between influenza A virus and sulfatide have potential as novel and more efficient antivirus drugs that can be used against conventional drug-resistant viruses or for patients with serious symptoms caused by advanced infection.
Sulfatide is a highly multifunctional glycolipid involved in the nervous system, diabetes, the immune system, hemostasis/thrombosis, bacterial infection and virus infection, as described in this review. Therefore, if sulfatide in the host cells is blocked or down-regulated by a direct binding agent, genetic technique or pharmacodynamic agent affecting sulfatide metabolism, it is very likely that many side effects associated with sulfatide will occur.
For treatment of influenza A virus infection, inhibitors specifically and directly targeting the sulfatide binding site on viral HA may be useful for preventing the predicted side effects and confer more powerful antiviral activity due to inhibition of progeny virus particle formation than NA inhibitors that inhibit the release process but not the virus particle formation process.
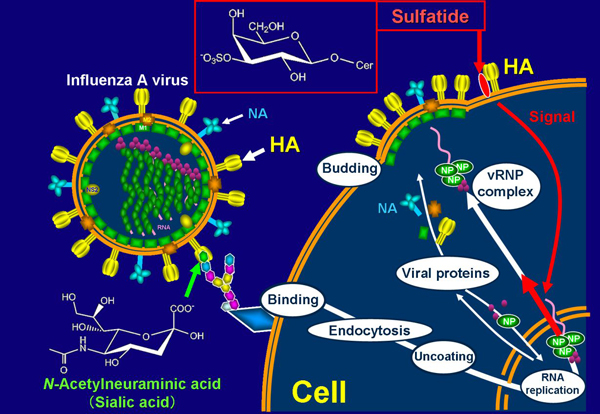
Fig. 3 Promotion mechanism of sulfatide on influenza A virus replication.
Influenza A virus binds to sialic acid, as a virus receptor, located in the terminal of glycochain on the host cell surface. The endocytosed virus particle then induces the fusion process to the endosome/lysosome membrane by acidic condition-triggered conformational change to fusogenic HA and release viral internal proteins and genomes to cytosol. Replicated viral RNA genomes in nucleus form vRNP complexes, together with newly synthesized viral NP and viral polymerases. vRNP complexes in nucleus are exported to cytosol. Newly synthesized viral envelope glycoproteins, HA and NA, are transported to the cell surface membrane. These proteins and other viral structural proteins form progeny virus particles through budding from the cell surface membrane. When sulfatide binds with HA transported to the cell surface membrane, it initiates unknown intracellular signals (intracellular narrow red arrow) that promote nuclear export of vRNP complexes (intracellular thick red arrow). This promotion results in increased formation of progeny virus particles.



