氏名:Andrew Almond
Andrew Almond博士は、1994年にEdinburgh大学で物理学の学位を取得後、Manchester大学School of Biological Sciencesの博士課程に進んだ。博士論文のテーマは、糖質(特にヒアルロン酸)と水との相互作用の理論的・実験的な研究を軸としていた。1997年にPh.D.を取得してからはWellcome TrustとBBSRCリサーチフェローとなり、そこでタンパク質存在下・非存在下におけるヒアルロン酸の動力学的コンフォメーション研究のための、理論的および実験的なアプローチの開発に取り組むうちに、彼の研究分野はやがて他の多糖類やオリゴ糖へと広がっていった。現在は、Manchester大学School of Biological Sciencesで講師を務めている。

氏名:Timothy E. Hardingham
Timothy E. Hardingham博士はBristol大学で生化学を専攻し、1968年に同大学にてPh.D.を取得した。その後はロンドンのKennedy Institute of Rheumatology (1968-1994)でHelen Muir博士とともにプロテオグリカンおよび関節軟骨について研究することで研究生活の大部分を過ごした。この研究でヒアルロン酸が、軟骨プロテオグリカンであるアグリカンの会合体形成において重要な要素となっていることを明らかにした。1978年には英国生化学会からColworth Medalを授与された。さらに、その後の変形性関節疾患に関する業績により、1989年にRoussel International Prize for Research on Osteoarthritis、1991年にはCarol Nachman Prize for Rheumatologyの共同受賞者となった。現在はManchester大学School of Biological Sciences附属のWellcome Trust Centre for Cell-Matrix Researchで、教授として軟骨組織工学の戦略を研究している。また、細胞外マトリックスにおけるプロテオグリカンとヒアルロン酸の機能についての研究も続けている。
細胞外マトリックスの他の成分と一線を画すヒアルロン酸(以下HA)。その独特なマクロ物性は、実験が容易で、すでに50年以上に渡って研究および言及されてきた。しかし、HAの微細構造の理解と、それがどのようにしてマクロ物性に変化していくのかに関してはまだはっきりとわかっていない。我々は本稿で、マクロ物理的挙動の新たな分析を概観し、それを局所的オリゴ糖レベルにおけるHAの挙動を決定する分子間相互作用に関する最新の証拠に関連づけてみる。
HAが初めて精製されて以来、その特性は、従来の生物物理学的分析法を悩ませてきた。それまでの分析法は、本来はタンパク質分析用に開発され、分析対象の特性が完璧なニュートン溶質になることを要求されたためである。溶液中のHAの挙動は、たとえ低濃度にあってもニュートン的とも「理想的」ともかけ離れているばかりか、研究を進展させるためにはSandy Ogston、Torvard Laurent、Endre (Bandi) Balazsそして後にはBob Clelandといった一流の研究者たちの頭脳を要した。彼らの研究こそが、HAの特徴的な性質を理解していくうえでの確固たる基盤となる、理論的・実験的な枠組みを確立した1-3。このシリーズのHascall, Laurentによる「ヒアルロン酸:その構造と物性」およびLaurent の「20世紀におけるヒアルロン酸研究—発展の系統樹—」を参照されたい。
明らかにされたHAの主な特性は下記の通りである。
これら初期の研究のほとんどは、光散乱法、浸透圧法、粘度法そして沈降法の生物物理学的な測定に基づいておこなわれた。それが非理想的挙動に対する一層の理解を深めると同時に、その他の様々な分析方法から得た結果とも合致する、HAの基本的構造モデルをもたらした。しかしながら、非理想性の要因となったコンフォメーションの分子の詳細が何であったかに関しては、様々な解釈の可能性が残っている。
HA挙動の解釈の1つに、溶液中のHA鎖の自己会合に基づいたモデルがあった4。HAの自己会合が、HAのいくつかの特性に対する答えを教えてくれる可能性はあるが、まずHA特性のすべての実験的分析と一致しているかどうかを確認することが重要であった。HAの鎖間自己会合が、その物理特性に主要かつ支配的な影響力を及ぼす可能性に疑う余地はない。例えば、まずHAが水中で強い自己会合を示した場合、不溶性になる可能性があると仮定できる;次に、希薄溶液中の鎖間会合は同分子内で起こっている可能性が高いため、HA分子ドメインを収縮する傾向があり、そして高濃度での鎖間会合は、異分子間に生じ、安定したゲルを形成する。自己会合の影響は、形成された鎖間結合の強度と安定性によって異なってくるが、(上述した)既知のHA特性を考えると、強固な鎖間会合が存在することと矛盾しているように思える。しかしながら、弱い短時間の相互作用が存在している可能性は残されており、HAの水溶液にそういった相互作用の証拠を確立していくことが重要であった。
1990年代後半になってから、Hardinghamと共同研究者たちは、溶液中の特性を分析するための新しい手法を開発することでHA研究を大きく前進させた。 この手法は、溶液中のFITC標識HAの側方並進拡散を測定する共焦点FRAPに基づいていた5-7 (Fig. 1)。この技術は、濃度、pH、イオン強度そして温度といった、HAの流体力学的挙動を左右するいかなる要素も見極めることができる。分子間相互作用の強弱を判断する上でこの技術が特に重要だったのは、高濃度溶液および希釈溶液のどちらにも対応できることであった。また平衡技法であるため、他の生物物理学測定に大きく影響する濃度勾配と移動境界がない。得られた結果は、高濃度におけるポリマーの絡み分子を反映する、良溶媒中のポリマーの自己拡散のために確立された経験的関係8に基づいて解釈することができる。
さらに、HAの分子ドメインへの影響は、同値寸法の球形としてのHAの流体力学的挙動を表すStokes-Einsteinの式を使ってモデル化が可能だ8。したがって、高濃度、低濃度の両方の自由溶液中の平衡状態におけるHAの流体力学的挙動と分子間相互作用の研究をおこなうのにまさに理想的であった。これにより、濃度が高くなるにつれてHA分子ドメインが「混雑し」重なり合っていく際に起こるHAの個別分子特性、分子間相互作用そしてネットワーク形成特性に幅広い特徴付けをすることができた。HAの自己会合を研究する上で後者がもっとも重要であったのは、弱い鎖間相互作用が関係している可能性があったからである。何故なら高濃度では鎖は溶液中で狭い間隔に並ぶはずで、たとえ微弱であっても鎖間の相互作用が存在していると十分に考えられたからであった。また共焦点FRAP技術はトレーサー拡散測定に適していたことから、高分子化合物の濃溶液が、より分子サイズの小さい、溶性のFITCで標識されたトレーサーの拡散を妨ぐのを分析することができた9。これは濃溶液中の高分子化合物によって形成された「ネットワーク」の見掛けの細孔の大きさの測定を可能にした。
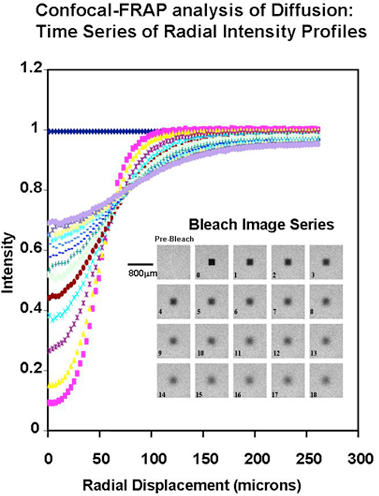
Fig. 1
拡散の共焦点FRAP分析:時系列でみる光量分布並進拡散の共焦点FRAPによる分析。FITC-デキストラン(分子量260 kDa、濃度100 μg/ml、25℃および中性pH 0.1 M塩化ナトリウム)での実験での共焦点画像と光量分布の一例。0時間での画像は、光退色3秒後のもので、続く各画像は6秒間隔である。(実験詳細はRef. 5を参照)
濃度の上昇に伴うHAの並進拡散における変化に対する初期分析は、溶液中の絡みによって表される特性を見せた(Fig. 2)。これは溶液中のすべてのポリマーに共通する特性である8。これは、水などの良溶媒に溶解したポリマーとしてのHAが、良溶媒中の他の高分子化合物と相当の挙動をとったことを示唆し、絡み以外に高濃度における分子間相互作用の証拠はなかった。イオン強度の影響が観察されたことも非常に有益であったのは、溶液のイオン強度を下げるにつれて、HAの流体力学的容積は大きな膨張を見せたからである(Fig. 3, 4)10。
これはHA鎖上で隣接するカルボキシル基の電荷反発の高分子電解質効果を反映したもので、これが流体力学的ドメインの拡大を引き起こす。この結果は、HAが高度に伸展した流体力学的ドメインを持った高分子化合物であることを立証し、過去に確立された硬いランダムコイルのモデルとも完全に一致していた。その絶対的な特性は、極めて分子量依存的であり、HAの明白な特性は2糖分子10,000にも及ぶその鎖の長さゆえ顕著であることを証明した。pH 4-8という広範囲では、HAの拡散挙動にほとんど変化は見られなかった。しかしながら、強アルカリ溶液(pH 12-14)では、流体力学ドメインが縮小したところを見ると、鎖を硬化させる要因は何であれ高pHでは失われたことが示唆され(Fig. 4)、実験時の状況下ではHAの解重合はほとんど見られず、pHを下げるにつれて元の性質に回復した6。アルカリの影響はおそらく、HAのいくつかのヒドロキシル基グループがイオン化していくことに起因すると考えられる。
興味深いことに、それはHA脱イオン水中の分子ドメインの1%未満にまで減らし(Fig. 3)、HAの特性は同様の分子量のデキストランのそれと類似するものに変化した。HAの自己拡散の温度依存性の研究は、とりたてて大きな成果は得られず、温度による水の粘度の変化と相関する拡散係数になめらかでわずかな変化を見せただけだった。結果は、鎖間会合があった場合に予想される、HAの分子間相互作用で生じる熱融解の証拠を示さなかった。HAオリゴ糖との競合実験においても、鎖間会合の形跡はなかった。鎖間相互作用を通してのHA分子の自己会合がHAの拡散を弱めるのに対し、短いオリゴ糖との結合が拡散率に影響するとは考えにくい。この場合、オリゴ糖は鎖間相互作用の競合物質となるはずだが、HA自己拡散にはまったく影響がなかった。いかなる変化も見られなかったことは、HA水力学的特性が鎖間競合に影響されなかったことを証明している9。
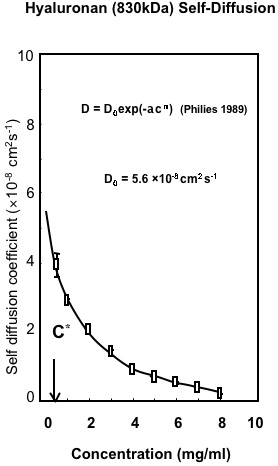
Fig. 2
HA (830 kDa) の自己拡散:HAの巨視的特性。共焦点FRAPによって決定された濃度のHAの自己拡散の変動。結果は、溶液中のポリマーのために提案された普遍的スケーリング方程式に合わせている8。

Fig. 3 HA (500 kDa)
脱イオン化水中におけるHA流体力学的ドメインの比較:
HA流体力学的ドメインへの塩と高pHの及ぼす影響。希薄溶液中の並進拡散係数に対するStokes-Einsteinの式の適用に基づいた、球状ドメインで表されている結果。濃度がゼロの時の拡散係数D0、ボルツマン定数κ、温度T、溶媒粘度ηそしてRhが流体力学半径のとき、D0 = κT/6πηRhである。
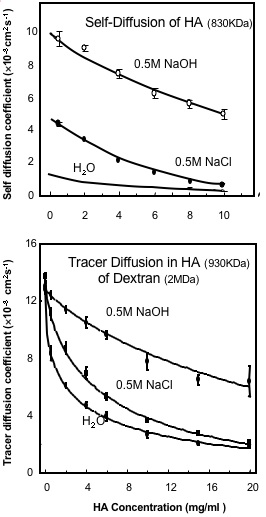
Fig. 4
塩と高pHが (A) HAの並進自己拡散係数と (B) HA溶液中のデキストラン (2 MDa) のトレーサー拡散に及ぼす影響6。HAは(A)では830 kDa、(B)では930 kDaである。
HA高濃度溶液中 (930 kDa と > 1 mg/ml) のFITC標識デキストラン(2 MDa)のトレーサー拡散を分析すると、拡散の濃度依存的な減少が見られ6、濃度を1 mg/mlから10 mg/mlに上げると、平均的細孔サイズである135 nmが30 nmにまで縮小するモデルを作ることができる。こうした特性は広いpH領域中(pH 4-8)ではほとんど変わらず、イオン強度でもわずかな変化しか見せなかった6。その理由は、鎖の濃度がイオン強度に対して不変であるため、トレーサー分子の拡散減少の主要な決定要因である溶液中の鎖密度に変化がないためである。イオン強度が低くなるとHA流体力学ドメインは拡張し、自己拡散が縮小するにもかかわらず、濃度がc*以上(個々の分子ドメインが重なり始める濃度)である限り、鎖密度への影響はない。濃度がc*以下のHA溶液が不均一なのは、分子ドメイン内に鎖密度が高い領域だけでなく、分子ドメインに鎖のない領域が存在しているためである。したがってc*以下のHA溶液のトレーサー拡散率は、HA分子ドメイン外の自由拡散率とHA分子ドメイン内の減少率の時間平均となる。c*以下の濃度になると、イオン強度がトレーサー拡散にいっそう大きな影響力を持つのは、分子ドメインはイオン強度が低いと増大し、高いと減少する性質を持っているからである。
もし細胞間マトリックス内のHA濃度がc*を上下するようなことがあると生理学的影響が出るだけでなく、HAの分子量の変化によってさらなる生理学的影響が生じると考えられる。もしHAが分解された場合には、その分子量と水力学的ドメインが縮小される。そうなるとドメイン重複のために必要な濃度、つまりc*は上昇する。したがって、たとえ濃度に変化がなくとも、分解が起こると高分子量HAに対してはc*より上に、低い分子量のHAに対してはc*より下の濃度に切り替えることができる。そしてこれは細胞間マトリックスのネットワークを通じて、HA自己拡散率および他のマクロ分子の拡散率を増大させる。こうして組織内HAの分解は、たとえその濃度の低下がなくとも、細胞から細胞へとシグナルを運んでいくマクロ分子のECMを通してアクセスを増加させる。
HAの分析に共焦点FRAPを使用するにあたり、一つの条件下でのみ自己会合を裏付けるわずかな証拠があり、それは何年も前になされた見解と密接に関連していた。かねてよりHAは、溶液がpH 2.5に調節されると粘弾性のある「パテ(putty)」を形成すると報告されていた11。この現象はこのpHに非常に特有であり、pH 3.0以上でもpH 2.0以下でも観測されなかった。共焦点FRAP分析では、pH 2.0およびpH 3.0と比較すると、pH 2.5では自己拡散の急激な減少が見られた。興味深いことに、pH 2.5ではHAのトレーサー拡散が増加するという現象が同時に見られた12。この2つの観測結果、つまり低自己拡散と高トレーサー拡散は、このpH条件下で鎖間会合が起こるモデルと一致している。なぜなら、鎖の会合は一時的に分子間結合を作って自己拡散を減少させるからである。さらに鎖間会合部位の形成は、不規則な鎖の局所分布を作り出すが、これによって平均より大きめの細孔サイズを生み出すため、トレーサー拡散が増大する12。pH 2.5におけるHAのパテ形成の分子的説明は不明だが、このpH条件下におけるカルボキシル基の部分的プロトン化がかかわっているのではないかと推測されている。しかし最も興味深いのは、予測された自己会合が共焦点FRAPにより検知されたことである。pH 2.5条件下で検知された通常と異なるHAの挙動は、より生理学的なpH条件下においても、またいかなるイオン強度あるいはHA濃度でも、類似の鎖間会合は存在しないことを確実に立証する手だてとなった。
このことからもわかるように、HAは普通の挙動を示す、大分子量ポリマーであり、純粋溶液中では硬いランダムコイルとして挙動し、分子の絡み合いによって決まる粘弾性溶液の形成の原因となる、極めて大きな水力学的ドメインを持っている10, 12。これらの特性は、その機能の一つとして大空間を占める水統御ポリマーとして作用し、同時に他の生物活性分子が複数列に組み立てられる多価の骨組みを提供し、タンパク質と多くの高度に選択的な相互作用に関与するという、HAの生物学的特性と一致している。
過去にHAの研究者は、実験結果から分子レベルにおいては、HAは完全に非拘束的なポリマーではないという結果に辿り着いた。なぜなら一つには、ポリマーは低濃度において粘弾性溶液を形成するが、これは自由に屈曲できるランダムコイルとは一致しないし、二つ目にはHAの過ヨウ素酸酸化は予測よりも、もっとゆっくりと起こるからである13, 14。しかし、多くの実験結果はHAには局所的な剛構造は存在しないとも示している。HAは高濃度まで可溶であり、凝固せず、弾性ゲルの形成に抵抗し、鎖間会合が見られない9。共焦点FRAP(上記参照)などの技術から得た最近の結果もこれを裏付けている。では、我々はどうすればこの2つの見解を両立させることができるのだろうか?そこで我々は、その3次元構造において平均的な局所構造秩序は持つものの、その平均構造の周囲でかなり動的屈曲を見せる分子によって、これらの結果を説明できると仮定してみた。我々はここでこの構造的仮説が、分子力学(molecular dynamics:以下MD)計算や、核磁気共鳴(nuclear magnetic resonance:以下NMR)による実験データの両方と一致し、同時にHAのバルク特性と、その生理学的挙動と機能の説明もできると主張したい。
HAの3次元分子構造を最初に垣間見ることができたのは、1970年代の配向線維のX線散乱からだった15。単一の、明確な配座よりむしろ、環境因子(例:イオンの種類、鎖の水和レベル)によって、回折パターンに劇的な変化が見られた。線維の回折パターンを分析することで、分子のらせん対称を決定でき、かつ、有益な事例の場合には分子の3次元構造の基本モデルの作成も可能となる。HAの場合には、0.8-1.0 nmピッチの2-、3-および4回軸の左巻きらせんが確認された(本シリーズSheehan and Almond、 ヒアルロン酸:静的、流体力学的および分子動力学的観点からを参照)16。大多数の場合、単位格子は平衡して一本鎖を包含するが、特定の条件下になると逆平衡配座で単位格子1つにつき、鎖が2本観測される。このことから次の一般的考察が導き出された。まず、単位格子の形がそこに存在しているイオンの種類に大きく影響されている可能性があるが、これは溶媒シェルが異なるイオンと係わり合いを持っているからではないかと結論された。二つ目としては、X線回折と一貫していたモデルには、糖ユニット間の分子内水素結合のはっきりした可能性が頻繁にあった。三つ目は、線維回折パターンのすべてが存在可能なモデルの構築に使用できるわけではない。これは低pHで観測された2回軸のX線回折パターンが、完全に精緻化されなかったことからも明らかである17。したがって、X線回折によって生成される精緻化された構造は、非生理学的条件の下で実施されており、結晶化条件のアーテファクトである可能性があるため、細心の注意と共に取り扱われるべきである。
文献によれば、3回および4回軸らせんは、最も広い範囲下で発見されているが、最も生理学的(水和化したナトリウムイオンの)条件が整っていると発見されるのは、ほぼ間違いなく4回軸らせんである18。これらのデータに基づき、溶液中で最も起こり得そうな配座は分子内水素結合を伴う3回もしくは4回軸の左巻きらせんで、HA溶液観察によって支持されている(水力学的観察から結論した、HA鎖の緩やかな過ヨウ素酸酸化と局部剛性など)。さらにHAの多様な結晶形態を観察した結果、HAは溶液中では局所的に動的である可能性を示し、このことはHAが高濃度でも粘弾性を保ち、切断可能なゲルを形成しないという事実に裏付けされている。
X線回折研究は、HAの可能な局所配座に対する重要な洞察を与えてくれる一方で、原子レベルでの情報を直接提供しないため、結局は限界がある;得られた分子像は静的であり、HAの生理学的機能に対する適切な理解には制限が生じる。こうした限界を克服する手だての一つとして、MDシミュレーションなどの近代コンピュータ手法の導入が挙げられる。こうした手法は、理論的(量子力学に基づく)もしくは実験的(分光学)手法から計算された、一般的な経験的パラメータを使って分子のポテンシャルエネルギーの計算を試みる。こうしたポテンシャルエネルギーを使用することによって、極小エネルギーをとる立体配座が計算できる。しかしながら、現実的な自由エネルギーは分子エントロピーを考慮することでのみ入手でき、それにはポテンシャルエネルギー表面に沿って分子を動かすMDシミュレーションを要する。さらに、HAのような糖質は水との強い分子レベルでの相互作用があることを、分子内水素結合力学を理解する上で明確に検討する必要がある。このようなシミュレーションは、たとえHA4糖類でさえ数千もの原子を考慮しなければならないため計算量を必要とし、可能となったのは比較的最近のことである。
基本的なコンピュータ計算では、X線回折研究によって発見された典型的な左巻きの形状が、糖鎖同士の結合のための最小エネルギー構造と一致することを示す19。このような形状では、どちらの単糖間の結合でも水素結合が存在することができる。これは局所エネルギーを低下させ、かつ構造を安定化させる役割を持つ。どちらの単糖間の結合部においても、ヒドロキシル基から水素結合が形成される(隣接する環状エーテル酸素とOH3あるいはOH4間で)。さらに1-4結合部では、(力の加わった)水素結合が隣接のカルボキシル基アミド基から形成される可能性がある。一方、1-3結合部では、カルボニル酸素とOH2位ハイドロキシル基の間で水素結合が形成されることがある。その結果として得られた低エネルギー構造は、(X線回折研究と比較して)比較的収縮したピッチの2糖を持つ、左巻き3もしくは4回軸らせんと一致する。そのような構造は水分子存在下の条件でMDシミュレーションをおこなう場合の基盤として使うことができる(Fig. 5)。
HAオリゴ糖のコンピュータモデルが、1滴の水に入れられ、生理的温度にまで加熱されると、水分子はHA分子の表面で水素結合し、分子内水素結合を除去する。具体的には、例えば、水分子はアミドとカルボキシル基の間に入り込む。その結果、直感に反して、分子は実際にさらに伸展する20。こうした水溶性シミュレーションでは、局所配座は2糖類を基本単位とした、平均で約4回軸らせんであることが発見された21。このような4回軸らせんは0.96 nmという平均的な2糖類の長さ(一つおいた次に隣接するグリコシド酸素原子の間)を有し、平均的な軸方向の伸長は0.84 nmである(Fig. 6)。この平均的構造はprotein data bank(http://www.rcsb.org/)からコード「2BVK」でダウンロードできる22。
さらに動的構造を数値化する上で、便宜上ここでは軽(水素)原子を介して定義されるグリコシド結合を調べることが役立つ。したがって、φ角はH1-C1-O-C×、ψはC1-O-C×-H×と定義される;他の重原子定義は120![]() を足すか引くかで求めることができる(下記参照)。この命名法を用いることで、1-3結合の平均的(φ、ψ)角度は(50.7°, 9.7°) であり、1-4結合は(47.9°, 8.0°)となる;どちらの結合タイプも(φ、ψ) = (50°, 10°)周囲の比較的狭い領域に(標準偏差±13°)限定されている。
を足すか引くかで求めることができる(下記参照)。この命名法を用いることで、1-3結合の平均的(φ、ψ)角度は(50.7°, 9.7°) であり、1-4結合は(47.9°, 8.0°)となる;どちらの結合タイプも(φ、ψ) = (50°, 10°)周囲の比較的狭い領域に(標準偏差±13°)限定されている。
分子内水素結合の頻度は、シミュレーションの時間ステップ幅を使い、水素結合をフィルターで検出するという、簡単なアルゴリズムによって監視することができる。相応のフィルターとしては、原子間距離0.35 nmで120°より大きな角度(D-H-A)を持つ電気陰性物質原子のドナー(D)とアクセプター(A)(この場合、いずれかがOあるいはN)である。シミュレーションの途中で2糖当たり4つの分子内水素結合が見られ、そのすべてが溶媒水分子と急速な(フェムト秒単位)交換をおこなっている(分子内水素結合内で時間の40%未満を費やす)と予測され、それは局所的な鎖の柔軟性の原因となる(Fig. 7)。発見された水素結合は、X線線維回折研究と基礎的分子モデリング(上述)によって定義されたものと類似している。繰り返すと、1-3結合部ではOH2はカルボニル酸素に、そしてOH4は隣接の環酸素へ結合し、1-4結合部ではアミンはカルボキシル基に、OH3は隣接の環酸素へとそれぞれ結合する。
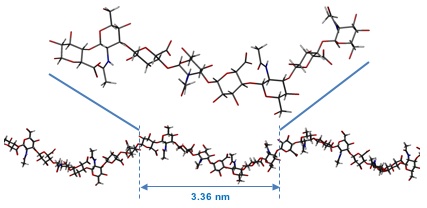
Fig. 6
水中のHAの分子力学シミュレーションから得られたHA 8糖の平均的な予測構造(上)。HAのより長いフラグメントの全体的な形状は、局所平均構造から派生したものである。このフラグメントは持続的に存在する長さよりも長く、平均構造を表しているものの、このような完璧な構造が起こるのは、溶液中で一瞬だけと予測される。
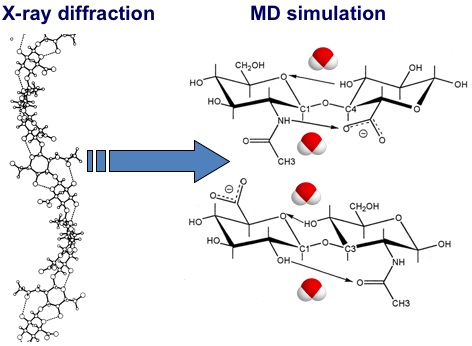
Fig. 7
固相でのX線回折研究と、分子力学シミュレーションから得た、HAモデルにおける水素結合の違い。静的X線回折モデルでは水素結合は偏在するが、分子力学シミュレーションにおいてはすべての分子内水素結合は、水分子への水素結合と競合状態にある。交換過程は迅速で(サブナノ秒)、分子内そして水分子を介した結合のどちらも、かなりの存在時間を持つ22, 25。
IUPAC命名法X線回折の文献に使われている)平均予測ねじれ角は次の通り:1-3結合 C1-O3-C3-C4は128.5°、O5-C1-O3-C3は-68.1°、1-4結合 C1-O4-C4-C5は-112.2°、そしてO5-C1-O4-C4は-71.1°である。この平均的コンフォメーションは、ナトリウムイオン存在下の線維から得られた斜方逆並行左巻き4回軸らせんとして決定されたX線回折像に近い。シミュレーションによって予測された2糖当たりの平均的な軸方向の伸長(約0.84 nm)は、固相HAで観察された最も収縮したらせんの一つである、これらのモデルと矛盾しない。回折パターンから得られたカリウムあるいはHAカルシウム線維はこの平均よりもさらに長く、それらは0.89-0.95 nm範囲の軸方向の伸長を持つ4回軸らせん鎖と、左巻き逆並行3回軸および4回軸らせん鎖を含む正方晶および斜方晶ユニットセルと特定された。こうした回折パターンから得られたコンフォメーションは、シミュレーションから予測された平均構造とは一致しないものの、前述の動的変動では精密に示されている。ただし、HAの生物学的性質の基盤であると定義されている軸方向の伸長0.98 nmの広がった2回軸らせんは過渡的にしか起きず、1-2個の2糖にわたる短いHA鎖に限定されている。
コンピュータモデルにより得られた、より生理的溶液条件下におけるHAの3次元情報の詳細な解析は、同位元素で標識したオリゴ糖のNMR研究から得られた。15N-原子核は(タンパク質などのように)オリゴ糖内での位置に敏感であり、すべてのアミド基はそれぞれが10糖と長さが同じ位のオリゴ糖内で同定することができた23。これは従来分析可能であった最大糖サイズ、4糖類(ポリマーとしてはあまりふさわしくないモデル)より大きな進歩である。オリゴ糖の部位特異的な位置において高感度緩和実験を実施することにより、結合角度はシミュレーションで13°と予測されていた標準偏差に近い約18°の標準偏差内にあることが推定できた24。さらに、NMRカップリング値の部位特異的測定は、trans (HN-N-C2-H2)から出るアセトアミド側鎖の回転運動の平均角度から約24°の標準偏差を示し、これもシミュレーションから推論された18°に近い(Fig. 8)。各アミド水素から隣接の環水素原子まで測定された、特定のNOESY法NMRのクロスピークもまた、以前に提唱された広がったた二重構造よりもむしろ、平均的なtransおよび収縮したHA局所構造のアセトアミド基と一致している22。
これらと同様に標識したオリゴ糖を用いることによって、オリゴ糖のどの位置からもアミドプロトン化学シフト摂法、カルボキシレート基pKa値、アミドプロトン温度係数および化学交換速度を確認することができる25。こうした観察は、アミド基とカーボキシレート基との間の高密度水素結合と一致しないことがわかっている。高分子HAとオリゴマーの特性を比較してみると、8糖類およびポリマーの中心とアミドプロトン環境との間には、明確な違いがないことが示唆され、やはりこれも3次構造の形成と矛盾している26。したがって、高密度分子内水素結合に依存する2次および3次構造モデルの重要な2つの側面は、微細的視点からは正しいとは考えにくい。
こうした結論は、アミド基の局所力学は近接のカボキシル基の存在に比較的影響を受けず、また1-4結合にわたるアミド基とカルボキシル基の間には、強く持続的な水素結合が存在する証拠が発見されなかったことを示唆するシミュレーションを反映している。その代わりに、この2つのグループはお互いに近接しており、局所的水分子と急速に交互置換する(強い相互作用は4%の時間でのみ存在した)弱い過渡的水素結合を通じて相互に作用し合うと推定される。この解釈は、上述した実験観察と一致しており、分子内水素が結合存在する時でも、時間の5-15%以上は存在できないことを示唆している。
こうした相互作用は、HAオリゴ糖に、外観上はほぼ4回軸だが、実質的な局所力学を有するコンフォメーションを維持する。水分子の関与はグリコシド結合により柔軟性をもたらし、比較的大きく、急激なコンフォメーション変化を可能にする。こうした解釈は、NMRとX線回折による微視的実験と一致しており、また、溶液中の硬くなったランダムコイルとしてのHAの流体力学的巨視的挙動とも一致する。
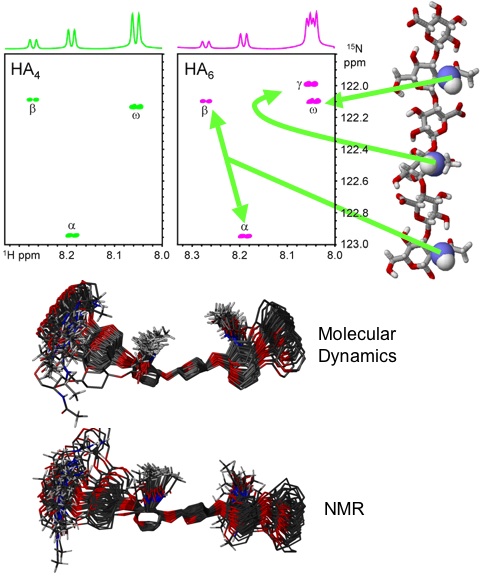
Fig. 8
HAの 15N-濃縮4糖および6糖の 1H-15N-HSQC NMRスペクトラが、全てのアミドプロトンは完全に分離されることを示している(上)23。水中におけるHA 6糖の分子力学シミュレーションに基づいたオーバーレイ(中央)が、15N標識HAオリゴ糖(下)のNMR実験による測定に基づいた予測構造の上に示されている22。
特異で独特なHAポリマー溶液の特性は、かなり以前に確立されたが、新しい手法によってそのマクロ高分子レベルにおける分子挙動をより明確に定義できるようになり、また局所および原子レベルにおけるその挙動を数量化・モデル化できるようになったのは、ごく最近のことである。そしてそれはHAの特徴的なマクロスケール特性が、その局所構造と周囲の水やイオンとの相互作用から、いかにして生じるかの明確な理解へとつながっていった。