氏名:野田 昌晴
自然科学研究機構・基礎生物学研究所
受容体型プロテインチロシンホスファターゼ ζ (Ptprz / PTPζ / RPTPβ)は主に中枢神経系に発現しており、コンドロイチン硫酸(CS)プロテオグリカンとして存在している(PG-B05参照)。最近、胃潰瘍の原因菌であるHelicobacter pylori(H. pylori)から分泌される細胞空胞化毒素(Vacuolating cytotoxin, VacA)の受容体がPtprzであることが判明した。
マウス及びヒトの組織を用いた解析により、胃粘膜組織におけるPtprzの発現量は脳の1/10以下ながら確かに発現しており、3種のスプライシングアイソフォームのうち、Ptprz-Bが主要分子であった。またPtprz-Bは胃ではCS修飾をほとんど受けていないことが判明した1。
VacAは細胞表面の受容体を介して細胞内に取り込まれた後、アニオンチャンネルを形成してエンドソームを膨潤させ、細胞内空胞を出現させることが知られている。マウスにVacAを経口投与すると、野生型マウスでは、全例において胃内に著しい出血を呈し、胃潰瘍が発症した。ところが驚いたことに、Ptprz-/-マウスでは、胃粘膜の障害が全く認められなかった(図 1)1。インドメタシンあるいはエタノール/塩酸の投与では、両マウスにおいて同程度に胃粘膜障害が生じたことから、Ptprz-/-マウスはVacAによる粘膜障害に対して特異的に耐性であるということになる1。
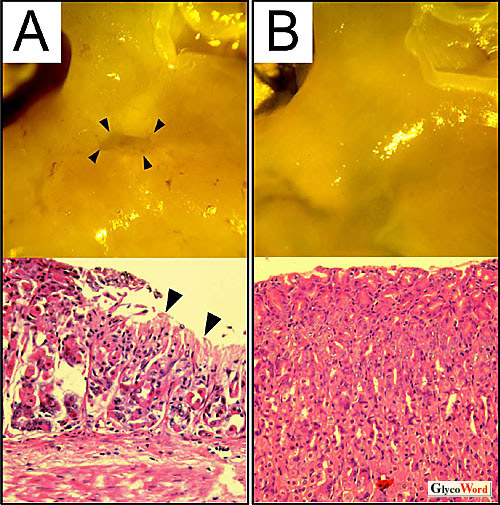
図 1 VacA経口投与後のマウスの胃
野生型マウス(A)では胃潰瘍が生じているが、Ptprz欠損マウス(B)には全く障害が認められない。
これまでVacAの細胞毒性に関しては、専ら細胞空胞化に起因すると考えられてきた。しかし、胃上皮細胞内へのVacA取り込みは、予想に反してPtprz-/-マウスでも野生型マウスと同様であったばかりでなく、胃上皮初代培養細胞を用いた実験から、VacAによって誘導される細胞内空胞化の程度も、Ptprz-/-マウスと野生型マウス由来の細胞で差は認められなかった1。このことは、細胞空胞化が胃潰瘍発症の本質的な原因ではないことを示唆している。
ところが初代培養細胞を用いた実験で、VacA添加によって野生型細胞が選択的に再構成基底膜から剥離する現象が見いだされた1。Ptprzの基質分子として、既に、細胞接着性や細胞骨格形成を制御する因子のひとつである、Git1が同定されている2。従ってVacAがPtprzのリガンド分子として作用したことが、細胞接着性に影響を与えた可能性が考えられた。Ptprzを発現させた細胞をVacAで刺激したところ、Git1のチロシンリン酸化レベルは、VacA刺激に対して用量依存的上昇を示した1。一方、Ptprz不活性型変異体を発現させた細胞ではGit1のリン酸化レベルは変化しなかった1。以上を総合すると、VacAが、Ptprzに結合してPTP活性を不活化した結果、Git1を始めとする細胞内基質分子のチロシンリン酸化レベルを亢進させ、これが胃上皮細胞−細胞外マトリックス間の接着障害を引き起こし、胃潰瘍発症に繋がっていると考えられる(図 2)。Ptprzの内因性リガンドであるプレイオトロフィン投与によっても、VacAと同様、野生型マウス選択的に胃粘膜障害が生じることは、この考えを支持する知見である1。
H. pylori感染は発癌の原因となることも指摘されている。VacAがPtprzを介してチロシンリン酸化シグナル伝達系に異常を引き起こすことは、発癌との関連性を想起させて興味深い。
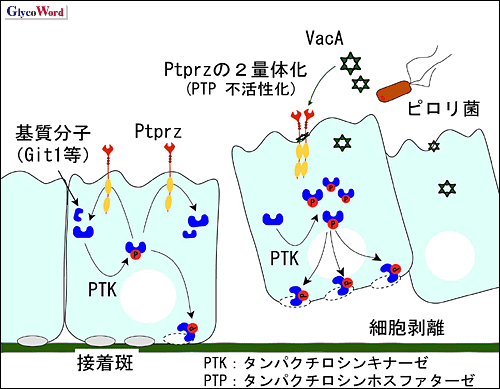
図 2 VacAによる胃潰瘍発生機序
VacAがPtprzに結合すると、Ptprz同士が会合し不活性化状態となり、その結果、基質分子のリン酸化レベルが亢進する。Git1のリン酸化は接着構造の分解に通じるため、細胞が剥離を始める。この細胞剥離箇所から胃酸等による組織障害が生じて、胃潰瘍が形成すると考えられる。



