
氏名:遠藤 玉夫
1977年東京大学薬学部卒業。1982年同大学院博士課程修了・薬学博士(指導教官、野島庄七教授)。米国Baylor医科大学博士研究員(Marcus教授)の後、1984年東京大学医科学研究所助手(木幡陽教授)。1994年(財)東京都老人総合研究所・糖鎖生物学部門室長。現在は組織改正に伴い、(財)東京都高齢者研究・福祉振興財団・東京都老人総合研究所・糖蛋白質研究グループリーダー(副参事研究員)。現在、老化、痴呆、発生における糖鎖の生物学的意義を明らかにしようとしている。
筋ジストロフィーは、筋肉が徐々に弱まり失われる遺伝子疾患である1。ここ15年の間にいくつかの筋ジストロフィーの原因遺伝子が同定された。その中でもっとも良く知られているデュシェンヌ型筋ジストロフィーは、ジストロフィンと呼ばれる蛋白質をコードする遺伝子の変異による。別のグループとして先天性筋ジストロフィーがあり、出生時あるいは出生後まもなくから筋の異常が認められる。最近特定の糖蛋白質、α-ジストログリカンの糖鎖異常が、ある種の先天性筋ジストロフィーの原因であることが分かってきた。本稿では、神経細胞遊走異常と筋ジストロフィーにおける糖鎖生物学の新知見を紹介したい。
ジストログリカンは一つの遺伝子でコードされており、蛋白質に翻訳後α-ジストログリカンとβ-ジストログリカンに切断される。骨格筋において、ジストログリカンは、ジストロフィン・糖蛋白質複合体(DGC)の主要構成成分のひとつである(図1)。α-ジストログリカンは細胞膜外表在性糖蛋白質であり、膜貫通糖蛋白質β-ジストログリカンに結合することにより膜に結合している。α-ジストログリカン・β-ジストログリカン複合体は生体に広く分布しており、細胞外マトリックスと細胞骨格を結ぶ連結軸として細胞膜の安定化に寄与していると考えられている。α-ジストログリカンは細胞外マトリックスのラミニンと結合し、細胞内ではβ-ジストログリカンがジストロフィンを介してさらにアクチン繊維と結合しているからである2。
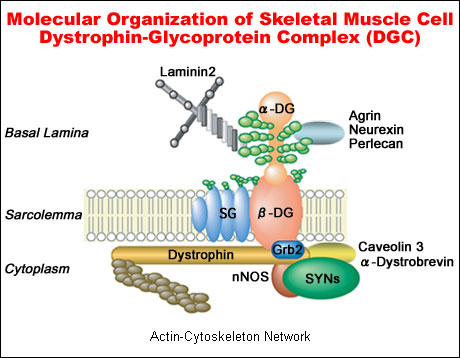
図 1
ジストロフィン・糖蛋白質複合体(DGC)と細胞外マトリックス成分ラミニンと細胞骨格成分アクチンとの連結軸の形成。α-ジストログリカンはDGCの中心成分でO-マンノース型糖鎖の修飾を受けており、糖鎖を介してラミニンと結合している。α-ジストログリカンは、ラミニンG領域を持つ細胞外マトリックスの他の成分であるニュ−レキシン、アグリン、パ−リカンなどとも結合する。一方、細胞内でβ-ジストログリカンは、直接あるいは間接的に様々な成分と結合する。DGC,ジストロフィン・糖蛋白質複合体; αDG, α-ジストログリカン; βDG, β-ジストログリカン; SG,サルコグリカン; nNOS,神経型NO合成酵素; SYNs,シントロフィン複合体。
α-ジストログリカンは糖含量の多い糖蛋白質であり、糖鎖はラミニン、ニューレキシン、アグリンとの結合に関与する3, 4。我々はシアル酸、それもO-結合型糖鎖上にあるシアル酸がラミニンとの結合に必要であることを明らかにした。そして、ウシ末梢神経α-ジストログリカンのO-結合型糖鎖の構造解析を行ない、ユニークなO-マンノース型糖鎖、Siaα2-3Galβ1-4GlcNAcβ1-2Manを発見した3。さらに、ウサギ骨格筋α-ジストログリカンも、やはり同じO-マンノース型糖鎖を持つことを明らかにした4。また、シアル酸を持つO-マンノース型糖鎖、Siaα2-3Galβ1-4GlcNAcβ1-2Manがα-ジストログリカン上のラミニンリガンドであることを明らかにした3。我々がシアル酸を持つO-マンノース型糖鎖の存在を報告した後、N-アセチルグルコサミンがマンノースの2位に結合するだけでなく2,6分岐のものや、様々な末端構造を持つ一連のO-マンノース型糖鎖構造 [Siaα2-3Galβ1-4GlcNAcβ1-2Man, Galβ1-4GlcNAcβ1-2Man, Galβ1-4(Fucα1-3)GlcNAcβ1-2Man, HSO3-3GlcAβ1-3Galβ1-4GlcNAcβ1-2Man, Siaα2-3Galβ1-4GlcNAcβ1-2 (Siaα2-3Galβ1-4GlcNAcβ1-6) Man] が報告された。現在のところ哺乳類におけるO-マンノース型糖鎖は、脳、神経、筋などの限られた糖蛋白質にみられる珍しいタイプの糖鎖修飾のようだ4。一方、O-マンノース型糖鎖は酵母では良く知られた糖鎖修飾であり、酵母の細胞壁にはマンノースが数残基つながったO-マンノース型糖鎖修飾を受けた糖蛋白質がたくさんある。今後の研究によって、哺乳類の組織でO-マンノース型糖鎖がどのような分布を示すか、あるいはα-ジストログリカン以外にどのような糖蛋白質がO-マンノース型糖鎖の修飾を受けているかなどが明らかになることを期待したい。
哺乳類におけるO-マンノース型糖鎖の生合成経路を明らかにすることは、O-マンノース型糖鎖の発現調節機構だけでなくO-マンノース型糖鎖の生物学的意義を理解するためにも重要である。哺乳類のO-マンノース型糖鎖の生合成に関わる酵素を同定しその性質を明らかにすることは、O-マンノース型糖鎖の理解に向けて重要なステップであろう。酵母型と哺乳類型のO-マンノース型糖鎖の大きな違いは、上述したように哺乳類型のO-マンノース型糖鎖はGlcNAcβ1-2Man結合を持つことである。この結合は、UDP-GlcNAcから糖蛋白質のO-マンノース残基にN-アセチルグルコサミニンを転移させる糖転移酵素、UDP-N-acetylglucosamine: protein O-mannose β1,2-N-acetylglucosaminyltransferase (POMGnT1)により形成されるのであろうと仮定した。そこでまずPOMGnT1の活性測定法を開発し、いくつかの動物脳で酵素活性を確認した5。ところで、GlcNAcβ1-2Man構造はN型糖鎖にすでに二つあることは良く知られており、UDP-N-acetylglucosamine: α-3-D-mannoside β-1,2-N-acetylglucosaminyltransferase I (GnT-I) とUDP-N-acetylglucosamine: α-6-D-mannoside β-1,2-N-acetylglucosaminyltransferase II (GnT-II) の二つの酵素によりそれぞれ形成される。しかしながら、O-マンノース型糖鎖上のGlcNAcβ1-2Man結合は、GnT-IとGnT-IIにより形成されるのではないことが分かり、新しい酵素の存在が示唆された。そこでヒトGnT-I遺伝子のホモローグを探しヒトPOMGnT1遺伝子を取得した6。ヌクレオチド配列からPOMGnT1は、推定分子量71.5 KDaの660アミノ酸からなる蛋白質であった。ゴルジ体に存在する他の糖転移酵素と同じくII型の膜蛋白質であると予想される。
ヒトPOMGnT1遺伝子は染色体1p33に存在することが判明し、まだ原因遺伝子が分かっていないMEB[MEB: OMIM 253280, OMIM = Online Mendelian Inheritance in Man (http://www.omim.org/)]の遺伝子座の近くにあることが分かった。MEBは、先天性筋ジストロフィーに眼奇形と脳(II型滑脳症)の形態形成異常を伴った常染色体劣性遺伝病である7。MEB患者はこのような重篤な症状を呈するが、成人に達することもある。患者はフィンランドに多い。DGC成分の欠損は筋ジストロフィーを起こすこと1, 2やα-ジストログリカンのO-マンノース型糖鎖がラミニンとの結合に必要であることなどから3、POMGnT1遺伝子変異とMEBには関連性があるのではないかと考えた。
この仮説を検証するために、MEB患者のPOMGnT1構造遺伝子とエクソン/イントロン配列について変異の有無を調べた。その結果、患者において6個の変異をみいだした6。その後さらに検索を進めた結果、7個の新たな変異をみつけ合計13個の変異をみいだした8。これらの変異は健常人ではまったくみつからなかったことから、これらの変異は病気の原因であり、POMGnT1遺伝子はMEBの原因遺伝子であるという結論に至った。これらの変異によりO-マンノース型糖鎖の合成が損なわれるかどうかを確かめるために、これらの変異体酵素を発現させ糖転移酵素活性を調べ、いずれも酵素活性が失われていることを明らかにした6,9。以上の結果より、MEBはPOMGnT1遺伝子の機能喪失による遺伝病であると結論した。もしPOMGnT1が機能しなければ、O-マンノース型糖鎖でみられる末端構造 (Siaα2-3Galβ1-4GlcNAc, Galβ1-4(Fucα1-3)GlcNAc, HSO3-3GlcAβ1-3Galβ1-4GlcNAc) はすべて形成されないことになる。これらの構造は接着過程に関わることが知られているので、O-マンノース型糖鎖の欠損は細胞移動や細胞接着に大きな影響を及ぼすことが考えられる。さらに、MEBではα-ジストログリカンの選択的な欠損がみつかった10。この結果より、α-ジストログリカンはPOMGnT1の標的分子のひとつであること、α-ジストログリカンの糖鎖異常はMEBの病態発症と大きな関係があること、を示唆していると考えられる。MEBでみられる筋と脳の異常は、O-マンノース型糖鎖の異常によるα-ジストログリカンの機能喪失によるのであろう。
最近の研究により詳細はまだ不明であるが、福山型先天性筋ジストロフィー(FCMD: OMIM 253800)、先天性筋ジストロフィー1C型(MDC1C: OMIM 606612)、Walker-Warburg症候群(WWS: OMIM 236670)、筋ジストロフィーモデル(myd)マウスなど他の筋ジストロフィーも、α-ジストログリカンの糖鎖異常によることが示唆されている(表1)。これらの異常を明らかにすることは、筋ジストロフィーの糖鎖病理機序の解明の新たな道を拓くことが期待される。
表 1 α-ジストログリカンの糖鎖異常が示唆される筋ジストロフィー
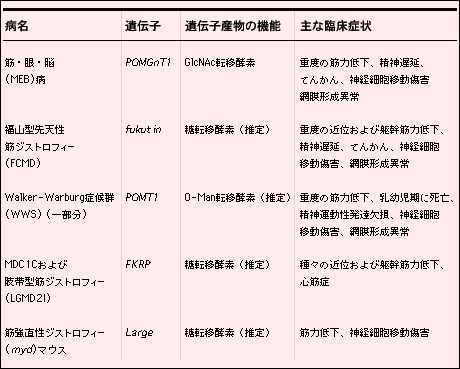
MEBと同じように、FCMDとWWSは先天性筋ジストロフィー、滑脳症と眼奇形を伴う常染色体劣性遺伝病である11,12。FCMDは日本において比較的良く見られる常染色体劣性遺伝病である11。日本ではデュシェンヌ型筋ジストロフィーに次いで二番目に多い子供の筋ジストロフィーである。発症頻度は人口100,000人当たり平均3人であり、日本では約90人に1人が保因者となる。小林らはFCMDの原因遺伝子を9q31に特定し、それはフクチンと名付けた461アミノ酸からなる蛋白質をコードしていることを明らかにした13。フクチンの機能はまだ不明だが、N末端にシグナル配列か膜貫通領域と思われる疎水領域を持っている。全体の配列は、細胞表面の糖蛋白質あるいは糖脂質を修飾する酵素を予想させる。最近武田らはフクチンの遺伝子を破壊した胎児性幹細胞を利用してキメラマウスを作製した14。このキメラマウスは、α-ジストログリカンを選択的に欠損しラミニン結合性を失っており、重篤な筋ジストロフィーを発症した。さらに、脳と眼の異常も示した。以上の結果より、フクチンは筋、脳、眼の正常な発達に必要であり、フクチンとα-ジストログリカンの密接な関係を示唆している。
WWSは、特に重篤な脳の形態異常、典型的なII型滑脳症、眼奇形を示すもう一つの先天性筋ジストロフィーである。WWS患者は出生時から重篤であり、ほとんど一年以内に死亡してしまう12。患者は世界中でみられる。最近、患者の20%(30人の患者中6人)でprotein O-mannosyltransferase 1 (POMT1)に変異がみつかった。POMT1は7つある酵母のO-マンノース転移酵素のホモローグであることから、セリンあるいはスレオニン残基にマンノースを転移する酵素であると予想されている15。POMT1は胎児脳、精巣、骨格筋というWWSで影響がみられる組織で特に高頻度で発現している。なおこれら30人の患者では、もうひとつのホモローグであるPOMT2の変異はまったくみつからなかった。このことは、この症候群では未知の原因遺伝子が他にあることを示している。ところで、POMT1 とPOMT2が実際にO-マンノース転移酵素であるかどうかは不明である。いまのところ脊椎動物のPOMTホモローグでO-マンノース転移酵素の活性の検出には成功していない。MEB と FCMDと同じように、WWSの骨格筋でもβ-ジストログリカンやラミニンは正常に発現しているにもかかわらず選択的なα-ジストログリカンの欠損がみられる3,10,15,16。ショウジョウバエでは筋肉の形成がうまくできないrt変異体というのが知られているが、この原因遺伝子はPOMT1のホモローグである17。rt遺伝子産物がO-マンノース転移酵素であるかどうかは不明であるが、O-マンノシル化は脊椎動物や無脊椎動物を問わず、筋の正常な発生に不可欠なのかもしれない。
これらに加えて、フクチンのホモローグ(fukutin-related protein, FKRP)の変異によって起こるMDC1Cでもα-ジストログリカンの糖鎖異常が観察される。MDC1Cは重篤な筋衰弱と変性、心筋症を特徴とする。精神遅延や小脳嚢胞が観察されることもある。FKRP遺伝子の対立変異は、しばしば心筋症を併発し、青年期から成人期と幅広い発症時期を示す肢帯型筋ジストロフィー2I型(LGMD2I: OMIM 607155)の原因ともなる18。FKRP遺伝子の変異を持つ患者は、例外なくα-ジストログリカンの発現が低下し、その程度は病気の重症度と一致(重症なものほどα-ジストログリカンの発現がより低下)し、さらにウェスタンブロット解析を行なうと糖鎖をより多く持つと思われる高分子量のα-ジストログリカンが明らかに減少していることが分かる。FKRPの機能は不明だが、糖転移酵素あるいは糖鎖修飾調節因子としてα-ジストログリカンの糖鎖修飾に関与すると考えられている。FKRPとフクチンはともにゴルジ局在蛋白質だと考えられるので19、これらの蛋白質の欠損がα-ジストログリカンの正常な成熟を妨げることは十分に考えられることである。
最後に、おそらく糖転移酵素遺伝子と思われるlarge遺伝子の変異による筋ジストロフィーモデルマウス(mydマウス20)を紹介する。しかしながら、この場合にもlarge遺伝子産物の酵素活性は証明されていない。mydマウスでの変異は、large遺伝子のエクソン5〜7の欠失である。この欠失は対応するmRNAのフレームシフトを引き起こし、未成熟な終止コドンを生じさせる。mydマウスは、進行性の筋ジストロフィー、眼の異常、さらに大脳皮質、小脳、海馬での神経細胞の遊走異常と基底膜の亀裂を特徴とする中枢神経系の異常を起こす21,22。mydマウスにおける筋と脳でのα-ジストログリカンの糖鎖修飾低下は、MEBとFCMD患者と同様に観察される。
以上まとめると、MEB、FCMD、mydマウスの筋細胞膜で観察されるα-ジストログリカンの低糖鎖修飾は、ラミニン、ニューレキシン、アグリンの結合性を弱める22。つまり、糖転移酵素と予想される遺伝子の変異によるα-ジストログリカンの糖鎖異常は、細胞外成分との結合異常を起こしMEB、WWS、FCMD、MDC1C、mydマウスで共通にみられる筋変性と脳の形態異常を引き起こしていると考えられる。言い換えると、α-ジストログリカンに糖鎖がきちんと付かないと、これらの疾患あるいはモデルマウスでみられる筋と脳の異常を起こすのであろう。しかしながら、これらの原因遺伝子が糖転移酵素だとしたら、POMGnT1以外は基質がまったくわかっていない(図2)。それぞれの酵素を同定し性質を明らかにすることは、脳の異常を伴う先天性筋ジストロフィーの分子レベルでの発症機構の解明に大いに役立つことが期待される。
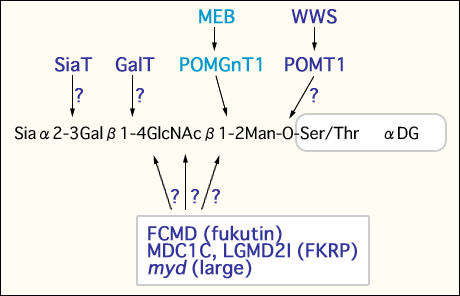
図 2 筋ジストロフィーにおけるα-ジストログリカンのO-マンノース型糖鎖の欠損(仮説)。
POMGnT1、POMT1、フクチン、FKRP、largeの変異は、α-ジストログリカンの糖鎖異常を起こす。しかしこれらの基質は、POMGnT1以外まだ分かっていない。POMT1は、O-マンノース型糖鎖合成開始酵素と予想されが、まだ証明されていない。ガラクト−ス転移酵素やシアル酸転移酵素の異常による筋ジストロフィーがあるかどうかは今後興味深い問題である。
ヒトの糖鎖合成欠損疾患は1980年に最初に発見され、その後今日まで10種類以上の先天性糖鎖形成不全症(CDG)が知られている。それらはI型とII型に区別されこれらはN型糖鎖形成不全に関するもので、詳細はFreeze博士により本シリーズで紹介されている23。しかしながら本稿で紹介したように、ある種の筋ジストロフィーはN型糖鎖合成には関与しない糖転移酵素の変異によると考えられる。これらの遺伝子産物の本質を解明することは、筋ジストロフィーという難病の解明に役立つことが期待される。筋、脳などで発現しているα-ジストログリカンの糖鎖異常は、これら筋ジストロフィーに共通する現象のようである。α-ジストログリカンは、神経細胞遊走異常を伴う筋ジストロフィーに対する糖鎖治療学という新しい分野を切り開くキーワードとなるであろう。



