
Tadashi Suzuki
Dr. Suzuki graduated from the Department of Biophysics and Biochemistry, Faculty of Science, University of Tokyo in 1992. Obtained a Ph. D. degree in 1997 (Advisors: Prof. Yasuo Inoue/Assoc. Prof. Yasufumi Emori). From 1997-2000 he was a post-doctoral fellow at the Department of Biochemistry and Cell Biology, State University of New York at Stony Brook. In 2000, he was appointed as a Research Assistant Professor. His major contribution was the identification of the genes encoding the cytoplasmic peptide:N-glycanase (PNGase) in budding yeast, as well as the cytoplasmic endo-β-N-acetylglucosaminidase (ENGase) in human. From December 2001, he serves as a researcher of the Precursory Research for Embryonic Science and Technology (PRESTO), Japan Science and Technology Corporation (JST). From February 2002 he is also an RCF Assistant Professor at the Undergraduate Program for Bioinformatics and Systems Biology (UPBSB), Department of Biophysics and Biochemistry, Graduate School of Science, University of Tokyo. There he has continued to work on structure and functions of deglycosylating enzymes, ENGase and PNGase.
Peptide:N-glycanase (PNGase; also called glycoamidase or N-glycanase) is a deglycosylating enzyme which acts upon asparagine-linked (N-linked) glycans on glycoproteins/glycopeptides. PNGases isolated from plants or bacteria have been used as powerful “tool” reagents for structural and functional studies of N-linked glycans on glycoproteins. Curiously enough, the physiological significance of this enzyme itself has long been undocumented until recently.
It has been known that the cytoplasmic PNGase activity distributes ubiquitously in eukaryotic cells. Past studies have revealed that the deglycosylating enzyme is implicated in proteasomal degradation of misfolded glycoproteins synthesized in the ER. This review will summarize the current knowledge of the structural/functional features of this enzyme. For complete review, see references 1, 2.
In eukaryotes, many proteins synthesized in the cytoplasm are translocated across, or integrated into, the membrane of the endoplasmic reticulum (ER), a central organelle for delivery of these soluble or membrane proteins to their respective destination. As this organelle is also responsible for monitoring proper folding/assembly of proteins, those with abnormal structure arising from mutations or metabolic damage can be eliminated. These proofreading and elimination system (“quality control system”) ensures cells to avoid the intracellular accumulation of nonfunctional proteins. In this system, proteins that fail to fold productively are retained in the ER by various lumenal chaperones, assisting their folding and appropriate subunit assembly. However, if some proteins maintain undesired structure despite the effort by chaperone system, they are destined for destruction by a mechanism known as an “ER-associated degradation” (ERAD)3. ERAD involves three successive steps: (I) sensing of aberrant proteins in the ER, (II) the retrotranslocation of substrates into the cytosol, and (III) degradation of them by ubiquitin-proteasome system 4. It has also been suggested that there is an evolutionally conserved system specifically dedicated for N-linked glycoproteins, named glycoprotein ERAD (GERAD) 5. Understanding the detail of ERAD/GERAD system is significant from not only for our understanding of this basic cellular mechanism but also for the medical application regarding the treatment of various human inherited or acquired disorders caused by the abnormality of this surveillance system.
The cytoplasmic PNGase activity was first suggested to be involved in the ERAD machinery by Wiertz et al. 6. Since then the ubiquitous enzyme conserved from yeast to human has been shown to be utilized in GERAD process 1. The deglycosylation reaction must achieve an efficient degradation of N-glycosylated proteins, since the bulky modification of amino acid side chain such as N-glycosylation limits the substrate access into the proteolytic chamber, i.e. inside the cylinder structure of the 20S proteasome. However, at least in budding yeast, the degradation of misfolded glycoproteins, though delayed, can be completed without the presence of this enzyme 7. This result clearly demonstrates the existence of PNGase-independent degradation for misfolded glycoproteins. Nevertheless, considering the current molecular model of the 20S proteasome, it seems unlikely that the efficient two-way traffic of N-glycosylated protein in and out of the central cavity of the 20S proteasome can occur with the bulky N-linked glycan attached to the protein/peptide throughout the process 8. Moreover, gene analysis as well as biochemical studies have suggested that there is no redundancy in this cytosolic enzyme 7,8, not as in the case with ubiquitin-ligases. It remains unclear how the proteasome achieves the degradation of misfolded glycoproteins without the help of PNGase-catalyzed deglycosylation.
The gene encoding the cytoplasmic PNGase (PNG1 in yeast) was first identified in S. cerevisiae 7. Extensive survey in sequence databases has revealed that this enzyme is conserved throughout eukaryotes. The enzymes in budding and fission yeasts have a highly homologous “core” sequence, while orthologues in higher eukaryotes, unlike those from yeast, have extended domains at both the N- and C-termini of the core domain 1,7. One of the interesting features of the PNGases is the presence of a "transglutaminase" motif in the most conserved region by which they have been proposed to belong to the transglutaminase-like superfamily 9. Transglutaminase is an enzyme that establishes covalent links between proteins by formation of amide crosslinks between the side chains of glutamate and lysine residues (Fig. 1). In most cases, proteins in the transglutaminase family possess a putative catalytic triad consisting of cysteine, histidine, and aspartate residues. Since the proteins in the transglutaminase superfamily are so far known to be responsible for either formation (transglutamindase) or hydrolysis (protease) of amide bonds, it is not surprising that the cytoplasmic PNGase, an amidase, has been defined as a third type of enzyme belonging to this superfamily. Indeed, the potential catalytic triad in this domain is conserved in all PNGase orthologues. It is also important to note that reducing reagents such as dithiothreitol are required for in vitro enzyme activity of the cytoplasmic PNGases 10,11. This observation is consistent with the assumption that they have a cysteine residue that acts as a critical nucleophile for its enzymatic activity. Furthermore, the extensive mutagenesis study using yeast PNGase has shown that these amino acid residues are indeed essential for enzyme catalysis, suggesting a common evolutionary lineage for the cytoplasmic PNGases and transglutaminases 12.
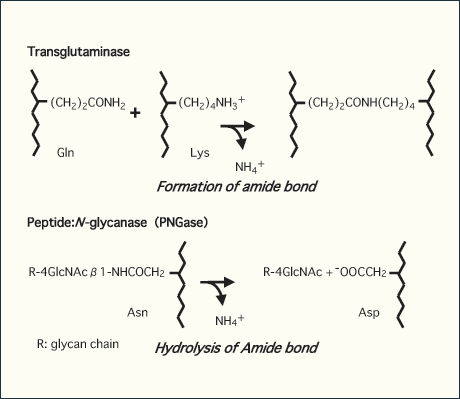
Fig. 1 chematic representation of enzyme reaction by transglutaminase (upper) and peptide:N-glycanase (lower)
While the transglutaminase catalyzes the formation of amide bond between side chains of Glu and Lys residues, PNGase catalyzes the hydrolysis of glycosylamide bond, forming a free oligosaccharide retaining di-N-acetylchitobioase structure at its reducing terminus as well as deglycosylated protein/peptide in which glycosylated Asn residue is converted to Asp.
We have shown that Rad23 protein (Rad23p) is a PNGase-binding protein both in budding yeast as well as in mammalian cells 13, 14. Rad23p, whose gene was originally identified as the one related with DNA repair, has been shown to exhibit multiple function in diverse cellular processes, while this protein is able to bind to the 26S proteasome by the N-terminal ubiquitin-like domain. In fact, the physical interaction of the yeast PNGase (Png1p) with the 26S proteasome occurs in a Rad23p-dependent manner in yeast 13. The Rad23p-Png1p complex has been found to exist as a distinct complex from the well-established Rad23p-Rad4p complex, which is required for DNA repair. Recent evidence also shows that the two UBA (ubiquitin-associated) domains in Rad23p can actually bind to ubiquitin/multiubiquitin chain, a major “tag” for degradation by the 26S proteasome. These observations suggest that Rad23 may serve as an “escort” function to connect the 26S proteasome with other proteins and exhibit diverse functions by controlling the stability and/or the functions of various binding proteins.
Regarding the complex formation of PNGase, the recent finding of the PUB/PUG domain as a novel sequence motif within the extended N-terminal region of PNGases from vertebrates and insects is particularly of interest 15, 16. This domain is highly homologous across the species, while PNGases from fungi or plants does not possess it. Our preliminary experimental evidence shows that the PUB/PUG domain is important for the binding of the mPng1p to a wide variety of ubiquitin/proteasome pathway-related proteins 14. Interestingly, all of the interactions except that of Rad23p orthologue, which requires the core catalytic domain for interaction, utilize the N-terminal domain, including the PUB/PUG domain, of mouse PNGase orthologue.
The functional importance of the PUB/PUG domain in proteins has not yet been proven. Nonetheless It is noteworthy that some of proteins that have PUB/PUG domains were found to have either a UBA or a UBX domain, a sequence motif present in multiple enzyme classes in the ubiquitin-related pathway, though the functions of these proteins are so far unknown. Recent studies suggest that both UBA and, most likely UBX domains function as protein-protein interaction domains 17. Accordingly, together with our experimental observation, it has been hypothesized that the PUB/PUG domain, as proposed for UBA or UBX domain, may also serve as a protein-protein interaction domain. One can readily envision that, together with UBA or possibly UBX domains, the PUB/PUG domain is able to generate a number of protein-protein interaction networks in many cellular processes including the ubiquitin-proteasome pathway. In this case, the acquisition of PUB/PUG domain may result in a formation of a “glycoprotein-degradation complex” in order to facilitate a series of reactions such as retro-translocation, ubiquitination, deglycosylation, deubiquitination and degradation of proteins at one cellular site 2. Demonstration of the occurrence as well as the characterization of these complexes would be the next logical step to substantiate this hypothesis.
The evidence that the cytoplasmic PNGase is involved in GERAD has been presented by several experiments. First, the small N-glycosylatable peptide, which follows the similar fate as that of misfolded glycoproteins, is deglycosylated by the cytoplasmic PNGase in the cytosol 18,19. Moreover, the amount of free glycan, the majority of which has been generated by PNGase in budding yeast 20, is dramatically reduced by inhibition of proteasomal proteolysis 21. This result indicates that the PNGase-mediated deglycosylation and proteasomal degradation are possibly correlated with each other. Furthermore, recent observation has proven that PNGase is capable of acting on misfolded glycoproteins that are GERAD substrates 8. This finding strongly supports the idea that PNGase deglycosylates glycoprotein prior to proteasomal degradation. The cytoplasmic PNGase seems to distinguish between native and unfolded status of glycoproteins, and only deglycosylates glycoproteins when they are extensively denatured 8. It is important to note that since the unfolding of proteins are not prerequisite for retrotranslocation of proteins from the ER to the cytosol 22, it is feasible to assume, at least in some cases, that there is an “unfolding” step of proteins prior to the action of PNGase to proteins in the cytosol.
The latest study has identified the lectin-ubiquitin ligase, Fbx2, to be involved in GERAD process 23. The lectin forms a SCF (Skp1-Cullin1-F-box) complex, which serves as an E3-ligase. It was revealed that the SCFFbx2 complex was able to recognize the misfolded glycoproteins for ubiquitination in N-glycan-dependent manner. Considering its reaction mechanism, the recognition of glycoprotein substrates by SCFFbx2 complex should presumably precede their deglycosylation by PNGase (Fig. 2).
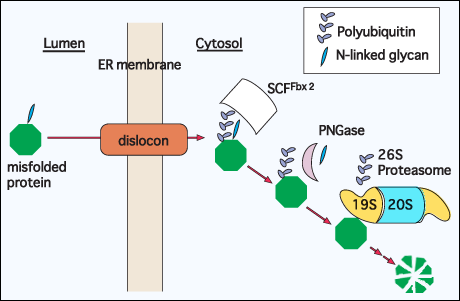
Fig. 2 Hypothetical model for degradation of a misfolded glycoprotein; cytosolic events
A glycoprotein that fails to acquire the correct structure is extracted from the ER using the novel protein channel, called dislocon. In the cytosol, it is recognized by a glycoprotein-specific ubiquitin ligase, SCFFbx2 complex, and ubiquitinated 23. PNGase then removes the N-linked glycan, and the polyubiquitin chain was removed by Rpn11 protein, a 19S proteasome subunit 25, 26, prior to the proteasomal degradation of protein.
When the occurrence of cytoplasmic peptide:N-glycanase (PNGase) was first reported in 1993 24, there has always been a difficult yet fundamental question regarding this enzyme; “Why in the cytosol?”. At that time few people believed that N-glycosylated proteins, the supposedly the substrates for the PNGase, can exist in the cytosol. However, after 10 years, it is now taken for granted that the “retrotranslocation” phenomena of (glyco)proteins from the ER to the cytosol does occur ubiquitously and today few people cast a doubt upon the implication of this enzyme in GERAD process, not to mention the presence of it. Now that the gene encoding the cytoplasmic PNGases has been identified and that structural features of this enzyme has been studied, the next focus will be on its functional aspect, especially in terms of phenotypic consequences due to its defect, in cells and individuals in higher eukaryotes.



