Jin-ichi Inokuchi
Hokkaido University
TNFα induces insulin resistance in type 2 diabetes but its mechanism of action is not fully elucidated. We have found that the expression of ganglioside GM3 and its synthase gene in adipocytes was upregulated by TNFα, and the increased GM3 suppressed insulin metabolic signaling through lipid microdomains. This observation may explain the new pathological aspect in lifecycle-related diseases such as type 2 diabetes.
Insulin elicits a wide variety of biological activities, which can be globally categorized into metabolic and mitogenic actions (Fig. 1). The binding of insulin to insulin receptor (IR) activates IR internal-tyrosine kinase activity. The activated tyrosine-phosphorylated IR was able to recruit and phosphorylate the adaptor proteins such as insulin receptor substrate (IRS). The phosphorylated IRS activates PI3 kinase, resulting in the translocation of glucose transporter 4 (GLUT-4) to plasma membrane to facilitate glucose uptake. This IR-IRS-PI3 kinase signaling cascade is the representative metabolic pathway of insulin (Fig. 1). On the other hand, the mitogenic pathway in insulin signaling initiates phosphorylation of Shc by the activated IR and then activates Ras-MAPK signaling (Fig. 1).
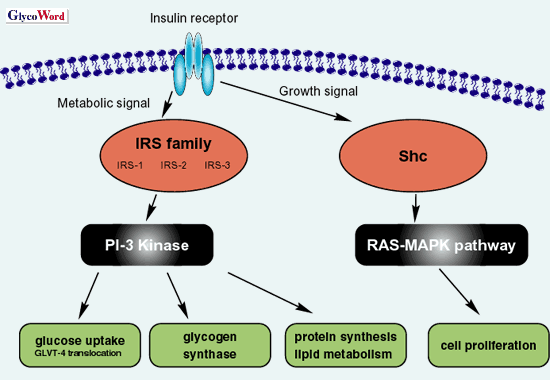
Fig. 1 Schematic diagram of representative insulin signaling.
In 1970, Berson and Yalow defined insulin resistance as "Decreased ability of cells or tissues to respond to physiological levels of insulin.” In the developed countries, pathological significance of insulin resistance is highly recognized as the cause of lifecycle - related diseases including hyperlipidemia, hypertension, hyperinsulinemia, and more significantly, obesity and type 2 diabetes. In the state of insulin resistance, the metabolic signals of insulin in liver, muscle and adipose tissues were selectively impaired. In fact, knockout mice lacking IRS-1 exhibited insulin resistance with hyperinsulinemia 1.
Adipose tissues from various species, including human, rat and mouse, contain GM3 as the most abundant type of ganglioside. It is known that, when GM3 is added exogenously to cultured cells expressing EGF receptor 2 and IR3, the inhibition of ligand-stimulated receptor autophosphorylation is observed. Moreover, there is a possiblility that endogenous GM3 may indeed regulate insulin signaling since IR exists in the membrane microdomains 4, the restricted membrane micro-compartment concentrating sphingomyelin, cholesterol and glycosphingolipids including GM3. We present evidence for the first time that the acquisition of a state of insulin resistance in adipocytes induced by TNFα may depend upon increased GM3 biosynthesis through upregulation of GM3 synthase gene expression and the GM3 may function as an inhibitor in insulin signaling during the chronic exposure of adipocytes to TNFα5. Here I describe our recent observation demonstrating that GM3 suppressed insulin metabolic signaling by affecting the functions of microdomains.
Insulin resistance in type 2 diabetes and TNFα
Genetically obese db/db mice, ob/ob mice and Zucker rats exhibit insulin resistance, and TNFα mRNA was expressed in their adipocytes at high levels compared to the control (lean) animals as well as obese diabetic human subjects. When the soluble TNF receptor-IgG kimeric protein was injected into Zucker rats to absorb TNFα, the state of insulin resistance was ameliorated. In addition, ob/ob mice lacking TNFα gene did not develop. From these observation, the considerable attention has been focused in TNFα over production by adipocytes accumulated neutral fats as the cause of insulin resistance.
Ganglioside GM3 as inducer of insulin resistance 5
When mouse 3T3-L1 adipocytes were cultured in low concentrations of TNFα which do not cause the generalized suppression of adipocyte gene expression including IRS-1 and GLUT-4, interference to insulin action by TNFα occurred. This requires prolonged treatment (at least 72 h), unlike many acute effects of this cytokine. The slowness of the effect suggests that TNFα induces the synthesis of an inhibitor that is the actual effector. We demonstrated that the state of insulin resistance in adipocytes treated with 0.1 nM TNFα was accompanied by a progressive increase in cell surface GM3. This was mirrored with increases in cellular GM3 content, GM3 synthase activity and GM3 synthase mRNA content, indicating that TNFα upregulates GM3 synthesis at the transcriptional level in cultured adipocytes. We were able to extend these in vitro observations to intact animals, since GM3 synthase mRNA contents in adipose tissues from the obese Zucker fa/fa rats and ob/ob mice were significantly high in comparison to the lean animals 5. We are currently analyzing the level of GM3 expression in adipose tissues from other rodent models as well as from humans with type2 diabetes.
To elucidate whether the increased GM3 in 3T3-L1 adipocytes treated with TNFα is involved in insulin resistance, we used an inhibitor of glucosylceramide synthase, D-PDMP (See Glycoword GL-46), to deplete cellular glycosphingolipids derived from glucosylceramide. D-PDMP proved able to counteract TNF-induced increase of GM3 content in adipocytes and completely normalize the TNF-induced defect in tyrosine phosphorylation of IRS-1 in response to insulin stimulation 5. These findings are supported by the recent unpublished observation that knockout mouse lacking GM3 synthase exhibits enhancement of insulin signaling6.
Regulatotry roles of microdomains in insulin signaling
Insulin-mediated internalization of IR into endosomes from plasma membranes and the intracellular movement of IRS-I to endosomes were significantly suppressed in 3T3-L1 adipocytes treated with TNFα, leading to the inhibition of IR-IRS-1 signalosome formation. Moreover, in the adipocytes-acquired insulin resistance by TNFα, IR accumulated less in the detergent-insobule low density membrane fractions (microdomains) and shifted to the high density fractions. Interestingly, under the state of insulin resistance, the IR – IRS-I signaling was selectively impaired without affecting the insulin-mediated MAPK activations. These observation strongly suggest that the defect of insulin’s metabolic signal in insulin resistance may be attributed to the loss of IR from microdomains and impaired endocytosis or IR(Fig. 2 and 3).
Further work is in progress in our laboratory to pursue the functional and structural changes of microdomains in the state of insulin resistance and type 2 diabetes to elucidate the fundamental roles of GM3 employing bioinformatics including proteome, metabolome and structural biology.
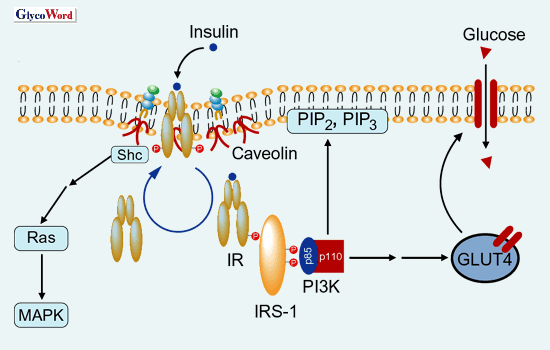
Fig. 2 Normal insulin mediated signal transduction of through microdomains
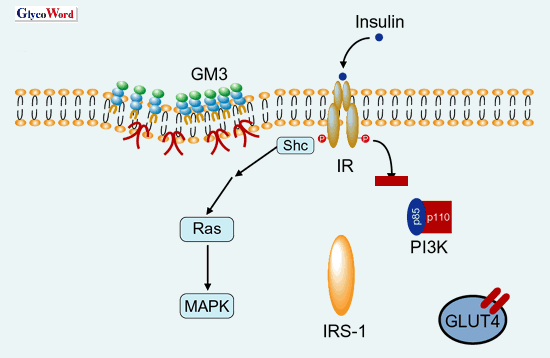
Fig. 3 Elimination of IR from microdomains and impaired metabolic action of insulin in the state of insulin resistance



