
Hudson H. Freeze
Freeze graduated from Indiana University with a BS in microbiology in 1968 and along with T.D. Brock discovered the original strain of Thermus acuaticus, the source of Taq polymerase. He then went to UCSD in La Jolla where he received his PhD in Biology in 1976. Postdoctoral appointments in Medicine and Neuroscience at UCSD led to an adjunct faculty appointment in Medicine 1983. He worked on the lysosomal enzymes of the slime mold Dictyostelium, and found their oligosaccharide structures resembled those on human lysosomal enzymes. In 1988 he moved to The Burnham Institute, then called the La Jolla Cancer Research Foundation, and was appointed full professor in 1994. His interest in Congenital Disorders of Glycosylation began when he noticed that oligosaccharides from fibroblasts of a CDG patient with an unknown defect had sugar chains similar to Dictyostelium glycosylation mutants. In the last several years Dr. Freeze discovered several new types of CDG and has encouraged physicians to agressively test for CDG patients. Since 2000 he has been the Director of the Glycobiology Program at the Burnham Institute, and continues to identify new patients and new types of CDG.
Essentially all secreted and cell surface proteins are glycosylated. The functions of the sugar chains are as varied as their complex structures, and include cell-cell communication, intracellular signaling, protein folding, targeting of proteins within cells and control of hormone levels, among many others. There are literally thousands of sugar chain structures. Unlike DNA and protein, which are linear molecules, the sugar chains are branched. Many sugars carry modifications….and then additional modifications are heaped onto them. Each one can significantly alter molecular shape and the ability to form higher ordered structures. The complexity makes their precise analysis difficult and initimidating. Studying sugar chain function and glycosylation is challenging. It has rarely been part of a typical biomedical graduate program, and essentially non-existent in medical education. Practicing physicians are seldom aware of it, and because there are few practicing Glycobiologists, physicians in training are not exposed to the field either. This is changing because we now know that altering glycosylation causes human disease.
Overview of N-Linked Glycosylation
All N-linked chains are derived from a common dolichol pyrophosphate (Dol-PP)-linked 14-sugar oligosaccharide (LLO). It is composed of 3 glucose (Glc), 9 mannose (Man) and 2 N-acetylglucosamine (GlcNAc) residues, Glc3Man9GlcNAc2, and then transferred en bloc from the lipid carrier to proteins. Each sugar is added to the growing LLO in a specific order using at least 13 glycosyltransferases. The oligosaccharide is then transferred to the nascent polypeptide chain by the oligosaccharyl transferase complex located in the ER membrane. After the transfer, the sugar chain is processed. Specific glycosidases trim the Glc3Man9GlcNAc2 chain, removing all Glc and some Man in the ER. Additional Man is often trimmed in the Golgi followed by the addition of 2-4 branches composed of GlcNAc, Gal and a terminal sialic acid (Sia) to form complex-type sugar chains.
Congenital Disorders of Glycosylation (CDG) 1,2
Congenital Disorders of Glycosylation (CDGs) result from defects in N-linked oligosaccharides that are added to Asn residues on nascent proteins in the lumen of the endoplasmic reticulum (ER) (Some of these are shown in Fig. 1).
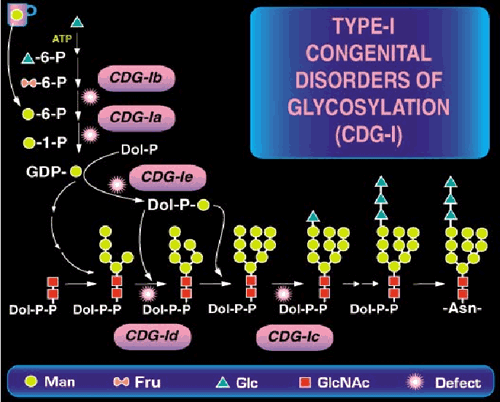
Fig. 1 A part of N-linked oligosaccharide biosynthetic pathway and location of the known defects.
During subsequent oligosaccharide processing many of these chains are trimmed and then extended once again with other sugars in various branching patterns. Since this is new frontier, few physicians think of glycosylation in diagnosing children with inherited diseases. Not surprising. Just five years ago we knew only two causes of CDG; now we know of 14 in just the N-linked pathway. Fig. 2 shows the dramatic explosion of new defects in the last few years.
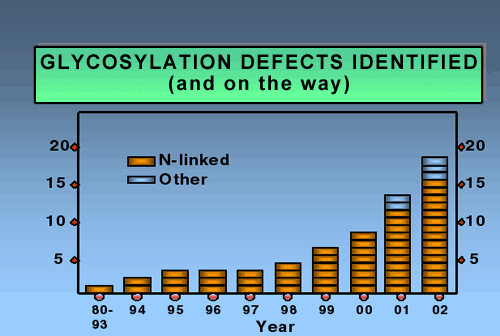
Fig. 2 Glycosylation defects identified
It is estimated that defects in any of the well over 50 genes needed for N-glycosylation will cause CDG, and many of these will likely be detectable by a simple blood test. Although there are currently about 300-400 cases of CDG world-wide, reliable estimates indicate this number accounts for only a few percent of the patients. It is safe to say that the entire group of CDGs is severely under-diagnosed, and that we are now only seeing the “tip of the iceberg”.
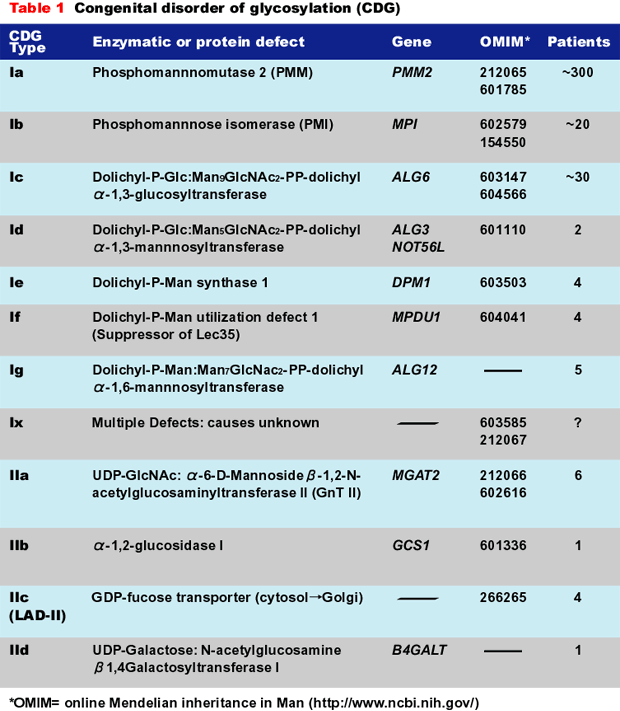
Table 1 shows the diseases, their causes, OMIM links, and number of known patients. Many of the patients were initially misdiagnosed. This situation is steadily improving, and that is important because two of the disorders (CDG-Ib and CDG-IIc) can be treated with simple monosaccharide therapy.
Biochemical Overview of CDG
The CDGs are autosomal recessive disorders. By far the most common type, CDG-Ia (OMIM 212066), is caused by mutations in the PMM2 gene. The gene encodes the phosphomannomutase used to convert Man-6-P to Man-1-P. Mutations reduce the size of GDP-Man pool and produce insufficient LLO for full glycosylation. Some of these patients have been mistaken for having mitochondrial disorders. CDG-Ib (OMIM 602579) results from mutations in the MPI gene encoding phosphomannose isomerase (PMI) (fructose-6-P→Man-6-P). The clinical pictures of CDG-Ib and CDG-Ia patients are quite different. CDG-Ic (OMIM 603147) is caused by mutations in ALG6, which encodes an α-1,3glucosyltransferase used to add the first Glc to the immature LLO precursor. Group II CDGs are defined as those that affect the processing of the protein-bound sugar chains, not LLO synthesis or its transfer to protein.
Laboratory Diagnosis of CDG
Glycosylation of serum transferrin (Tf) is used to biochemically diagnose CDG. Abnormal Tf is detected by isoelectric focusing, or by electrospray ionization-mass spectrometry. These analyses provide clues to the defect but neither one can pinpoint the gene.
Common Clinical Features of CDG
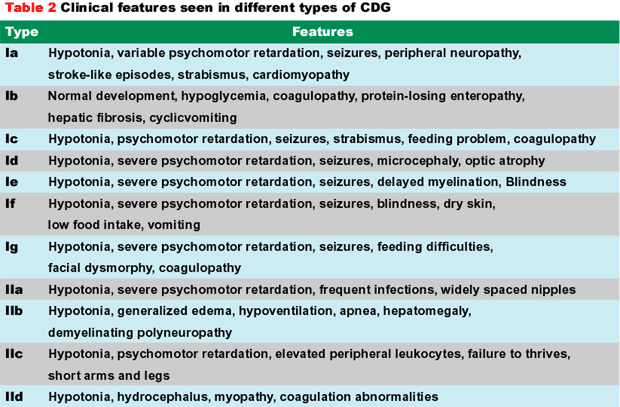
The most common clinical features in various types of CDG are shown in Table 2. There is considerable clinical heterogeneity. Psychomotor retardation ranging from mild to severe and hypotonia are consistent features in all patients except Type-Ib. Other neurological findings include ataxia (Ia and Ic), seizures and stroke-like episodes. Cerebellar hypoplasia (Ia, Ic), delayed myelination (Ie, IIa, IIb), microcephaly and atrophy of the cerebrum (Ia, Ic, Id, Ie) are seen. Sometimes the cognitive deficiency can be mild. Nearly all patients have feeding problems and fail to thrive. Strabismus, abnormal fat distribution, and retracted nipples are common.
Mortality in CDG-Ia children is about 20% during the first few years, but they stabilize after childhood. Substantial survival means that many adult CDG patients remain undiagnosed.
Limited Therapy Options for CDG
The effective therapy for CDG-Ib is oral mannose.3 Mannose by passes the fructose-6-P→Man-6-P block to replenish the depleted GDP-Man pools. Mannose reverses hypoglycemia and deficient anti-thrombin III within a few weeks, and within 1-2 months plasma protein levels are normal and protein-losing enteropathy disappears. None of the adult patients with proven CDG-Ib is currently taking mannose, suggesting that it will not be a lifelong requirement. One CDG-IIc patient was treated with fucose supplements that corrected his elevated circulating neutrophil counts by providing for Sialyl Lewis X synthesis.4
A key molecule on the surface of muscle cells is a-dystroglycan, which is part of the dystrophin complex that bridges the extracellular matrix and cytoskeleton.α-Dystroglycan contains a special type of mannose-based sugar chain, whose study was long neglected (except in Japan!). These sugar chains are not part of the N-linked pathway, but the chains are clustered together in small region of α-dystroglycan where they mediate many of the critical stabilizing interactions with the matrix. In hindsight now, it is not so surprising that several kinds of muscular dystrophies result from mutations in the genes needed for the biosynthesis of these sugar chains. Muscle-eye-brain disease, Fukuyama-type congenital muscular dystrophy (FCMD), and Walker-Warburg Syndrome involve mutations in these genes. Studying the glycosylation of α-dystroglycan in all its forms in many tissues and cell types will be important to understand how these conditions differ from each other. Even more surprising is the discovery that one form of adult onset muscular dystrophy, hereditary inclusion body myopathy-Type II, is due to mutations in an enzyme in the biosynthesis of CMP-Sialic acid, the universal activated donor for one of the sugars in the α-dystroglycan sugar chain.
The very complexity of sugar chains and their assembly practically guarantee that more disorders will be discovered. The most likely new areas are O-GalNAc-linked sugar chains, glycosaminoglycans, and Golgi assembly and organization proteins. These causes may be much more difficult to identify because many of these pathways are composed of redundant, sometimes overlapping enzymes with tissue specific distributions. Good examples are the multiple hereditary exostoses (MHE) in heparan sulfate synthesis, and the selective expression of the multiple fucosyl transferases that synthesize Lewis-X and Lewis-Y sugar chains. In comparison, finding defects in the early portion of the N-linked pathway has been easy because it is mostly a linear sequence with only a single enzyme at each step. Moreover, transferrin provides an unusually robust assay for defective N-glycosylation. The other pathways lack a similar diagnostic champion.
Glycobiology was called a Cinderella Science in 2001. Now, with the explosion of inherited glyco-pathologies, the pumpkin coach may become an ambulance, especially if we continue to find glycosylation-based therapies. Understanding the role of sugar chains in building cell-surface signaling complexes and finding how to boost sugar donors in cells may help us to understand and treat these pathologies. Many more defects will be found in the future.
Acknowledgements
The author is supported by grants from the National Institutes of Health, The March of Dimes Foundation and the CDG Family Network Foundation.



