



Biosynthesis of the common tetrasaccharide sequence in the glycosaminoglycan-protein linker region | |||||||||||||||
 |
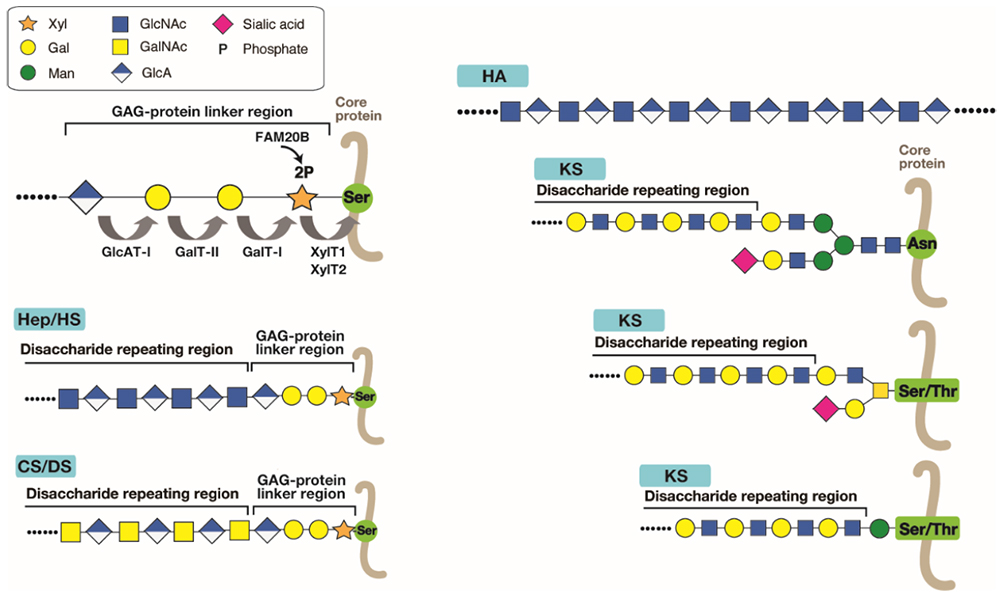
Glycosaminoglycans (GAGs) forming the proteoglycans (PGs) are the most abundant heteropolysaccharides in the body, and roughly classified into chondroitin sulfate (CS), dermatan sulfate (DS), heparin (Hep), heparan sulfate (HS), and keratan sulfate (KS). Hyaluronan (HA) is the only GAG type which is not sulfated, as well as lacking attachment to a core protein to form a PG (Figure 1) (see the section “Hyaluronan Synthase”). The glycan portions of PGs have distinct structures and functions. The GAG chains, except for HA and KS, are covalently bound to the hydroxyl group of the particular serine (Ser) residues of the core proteins through a tetrasaccharide linker (GlcAβ1-3Galβ1-3Galβ1-4Xylβ1-, wherein GlcA, Gal, and Xyl are glucuronic acid, galactose, and xylose, respectively) that is common to different GAG species (Figure 1). This surprising finding was made in the 1960s by a structural comparison of neutral glycopeptides isolated after enzymatic digestion and partial acid hydrolysis (1). KS chain is linked to the core protein via N- or O-linked glycans (Figure 1) (see the section “Biosynthesis of Keratan Sulfate and its Sulfation Steps”). Since the linker tetrasaccharide is common to CS, DS, Hep and HS, a failure to synthesize this structure leads to the loss of these GAG chains. The linker tetrasaccharide is formed by the sequential stepwise addition of monosaccharide residues by the respective specific glycosyltransferases, xylosyltransferase (XylT), β4-galactosyltransferase I (GalT-I), β3-galactosyltransferase II (GalT-II), and β3-glucuronyltransferase I (GlcAT-I) (Figure 1) (2). The transfer of β-Xyl from uridine diphosphate (UDP)-Xyl to the specific Ser residue(s) on the core proteins of PGs is mediated by XylTs encoded by XYLT1 or XYLT2. The second Gal is then transferred from UDP-Gal to Xylβ-O-Ser by GalT-I, encoded by B4GALT7. The third Gal residue is added to the second Gal residue on the Galβ1–4Xylβ-O-Ser from UDP-Gal by GalT-II, encoded by B3GALT6. During the formation of the trisaccharide linker, GAG Xyl kinase, encoded by FAM20B, phosphorylates the C2-position of Xyl using adenosine triphosphate as a phosphate-group donor. GlcAT-I, encoded by B3GAT3, transfers GlcA to Galβ1–3Galβ1–4Xylβ-O-Ser from UDP-GlcA. Concomitant with the transfer of GlcA to the linker trisaccharide, a phosphate group in GlcAβ1–3Galβ1–3Galβ1–4Xyl(2-O-phosphate) is removed by 2-O-phospho-Xyl phosphatase encoded by PXYLP1.
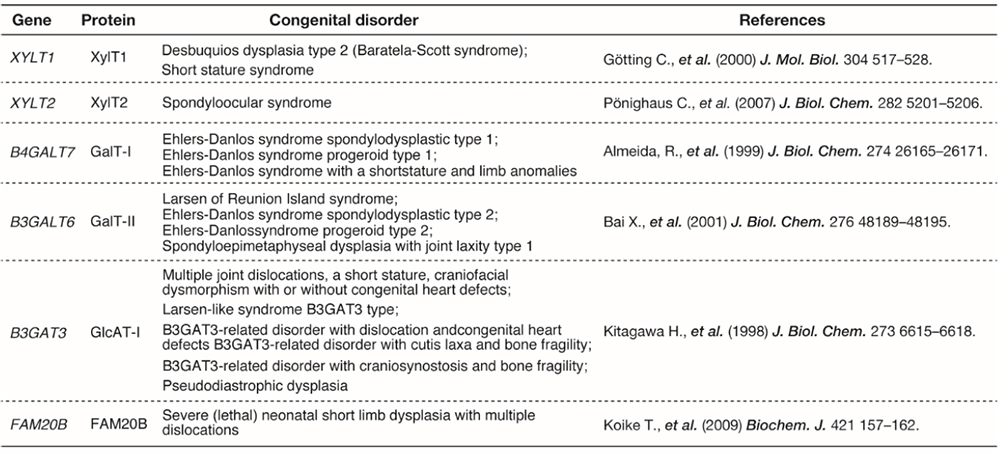
One of the major unsolved problems in GAG biosynthesis is the molecular mechanism for biosynthetic sorting of GAG species, including CS, DS, Hep, or HS. The amino acid sequence flanking the GAG attachment site of a number of PGs affects considerably the selective assembly of HS or CS on the GAG-attachment sites. For example, acidic amino acid clusters occur commonly in HSPGs and less frequently in CSPGs. Hydrophobic amino acids in close proximity to the Ser-glycine attachment site have been proposed to be an enhancer element for HS assembly. In addition, there is the possibility that sulfation and phosphorylation status of the linker tetrasaccharide controls GAG biosynthesis. However, the validity of this hypothesis has not been confirmed. Because sulfation of linker region has never been observed for HS/Hep chains, this modification may make a PG core protein bypass the biosynthetic route of HS. Furthermore, the biosynthetic enzymes physically interact within a macromolecular complex called GAGOSOME. The GAGOSOME model proposed in 2002 by Jeffrey Esko and Scott Selleck proposes that some of the HS biosynthetic enzymes may act together in a complex (3). Various GAGOSOMEs with different combination of biosynthetic enzymes coordinate polymerization and fine structural modifications of GAG in the Golgi apparatus. Several genetic disorders are caused by mutations in the genes encoding glycosyltransferases responsible for the synthesis of the tetrasaccharide linker (Table 1) (4). Diverse connective tissue disorders caused by mutations in these genes are collectively known as “proteoglycan linkeropathy” (5).
Satomi Nadanaka / Hiroshi Kitagawa
|
||||||||||||||
| References | |
|---|---|
| (1) | Lindahl U, and Roden L. In Glycoproteins, 2nd ed., Elsevier, New York, 491-517, 1972 |
| (2) | Mikami T, and Kitagawa H: Sulfated glycosaminoglycans: their distinct roles in stem cell biology. Glycoconj. J. 34, 725-735, 2017 |
| (3) | Esko JD, and Selleck SB: Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem. 71, 435-471, 2002 |
| (4) | Mizumoto S, and Yamada S: Congenital disorders of deficiency in glycosaminoglycan biosynthesis. Front. Genet. 12, 717535, 2021 |
| (5) | Nakajima M, Mizumoto S, Miyake N, Kogawa R, Iida A, Ito H, Kitoh H, Hirayama A, Mitsubuchi H, Miyazaki O, Kosaki R, Horikawa R, Lai A, Mendoza-Londono R, Dupuis L, Chitayat D, Howard A, Leal GF, Cavalcanti D, Tsurusaki Y, Saitsu H, Watanabe S, Lausch E, Unger S, Bonafe L, Ohashi H, Superti-Furga A, Matsumoto N, Sugahara K, Nishimura G, and Ikegawa S: Mutations in B3GALT6, which encodes a glycosaminoglycan linker region enzyme, cause a spectrum of skeletal and connective tissue disorders. Am. J. Hum. Genet. 92, 927-934, 2013 |
Dec. 13, 2023

