
氏名:細谷 隆史
京都府立大学大学院生命環境科学研究科准教授
経歴:2003 年3月京都大学農学部生物機能科学科卒業、2008年5月京都大学大学院エネルギー科学研究科エネルギー社会・環境科学専攻博士課程修了、JSPS特別研究員、ウィーン農科大学博士研究員,京都府立大学大学院生命環境科学研究科特任講師、同助教を経て2019年10月より現職。
専門分野:バイオマス化学、有機物理化学
グリコシド結合の化学合成(グリコシル化)は、様々な配糖体や多糖類の化学合成を可能にするための鍵となる反応である。グリコシル化は、糖の1位炭素に種々の脱離基が導入されたグリコシルドナーとアクセプター分子の間の求核置換過程である。求核置換反応は、大学初頭レベルの有機化学に登場する反応であり、すでに確立されたその制御法が存在するように思われる。しかしながら本反応は、求電子種であるドナーが多種の成分より構成される平衡混合物を形成するなど、通常の求核置換反応とは異なる複雑な過程を経て進行する。この複雑さゆえに、グリコシル化反応の制御には合成化学者の経験と勘に頼った試行錯誤が必要とされ、このような現状を打開すべく本反応における分子機構の解明が多くの研究者によって進められている。本稿では、グリコシル化の反応機構解明における鍵であるイオンペア類の化学特性の評価に主眼を置いて、該当分野における諸研究を紹介する。
グリコシド結合の化学合成、すなわち有機合成化学的グリコシル化は、糖化学の中で主要な研究分野の1つである。本反応は、図 1に示すように、脱離基Xを有するグリコシルドナーとアルコール、アミン、オレフィン等のアクセプターとの間の求核置換反応である1-5。特に、アクセプターとしてアルコールを用いたグリコシル化反応(O-グリコシル化反応)は、種々の配糖体、オリゴ糖、多糖類の人工合成に不可欠な反応であり、有機化学のみならず生化学、医学などの様々な学問分野で極めて重要な位置づけにある。著者の専門である木材化学の分野でも、木材の主要構成成分であるセルロースやヘミセルロースの諸性質を、化学構造の観点から系統的に調べる際などに、グルコシル化を用いた種々のモデル糖の合成が行われている6,7。
グリコシル化のこのような重要性にも関わらず、この反応は有機合成化学的な視点から確立された反応とはいえない。合成化学者が思い描く望みの糖分子の合成の成否は、化学者の経験と勘、そして何より払われた労力の大きさに依存する。この反応が抱える課題の中でも、特に大きなものは立体選択性(α/β選択性)である。1つの糖分子の合成条件の確立には、望まない方の異性体の副成を抑制するために、温度や溶媒といった比較的単純なものから脱離基Xやドナー水酸基の保護基の種類など複雑なものまで、多岐に渡った反応条件を細心の注意を払いながら1つ1つ設定する必要がある。そこに潜む困難さは、例えばたんぱく質や核酸類の人工合成法が確立されているのに対し、オリゴ糖や多糖分子の人工合成法の確立は未だ達成されないことからも理解されよう。
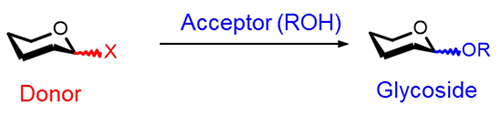
多くの課題が存在するグリコシル化反応であるが、こういった現状の一因は、種々の合成条件を理性的に検討する上での拠り所となるはずの、グリコシル化における機構論的知見が不足していることである。図 1に掲げた求核置換反応は大学の学部初頭レベルの有機化学であるが、この描像だけでは、本反応の本質は理解できない。反応機構をグリコシル化制御法のベースとするために、より微視的な視点に立った詳細な機構論的知見が必要である。
図 2には、現在広く研究されている脱離基Xにトリフレート(CF3SO2, TfO)基が採用されたドナー分子において信じられているグリコシル化機構を示している5。グリコシルトリフレートは1位炭素(C-1)とTfO間に共有結合を有する化合物であるが(Covalent Triflate: CT)、CTは反応液中で真の求核剤として働く化学種のごく一部に過ぎないと考えられている。CT中のC-1─OTf結合は、グリコシル化における反応溶媒として汎用されるジクロロメタン等の非極性溶媒中でも、自発的にイオン解離する。これはTfO−の高い安定性だけでなく、イオン解離によって糖側から生成するオキサカルベニウムイオンのC-1カチオン中心が、O-5からの電荷移動によって高度に安定化されることによる8。C-1とTfO間の共有結合のイオン解離が進行すると、静電相互作用により両者が緩やかに結合した緊密イオン対(Contact Ion Pair, CIP)が生成する。CIPからさらにイオン解離が進むことで、溶媒分離イオン対(Solvent-Separated Ion Pair, SSIP)が生成する。CT、CIP、SSIP間の相互変換における活性障壁は十分に小さいと考えられるため、グリコシルトリフレートは、反応系で生成した瞬間にCTやイオンペア種より構成される平衡混合物を形成することになる。
CT、CIPおよびSSIPは、どれも図 2でROHと記載されているアクセプターと反応しグリコシドを与えるが、その際の反応様式は各求電子種によって異なる。共有結合性のCTは、C-1周りの立体反転を伴うSɴ2機構でアクセプターと反応する。つまり、CTのα-アノマー(αCT, 図 2)からはβ-グリコシドが生成し、逆にβCTからはα-グルコシドが生成する。一方SSIPの反応では、アクセプターとのSɴ1-likeな反応が進行することで、α体とβ体の両方のグリコシドが生成すると考えられている。CIPの求核置換反応は、CTとSSIPの中間の機構で進行するとされるが、一般にTfO−が存在する面とは逆の面からのアクセプターがドナー分子に攻撃するSɴ2-likeな反応が進行すると理解されている。
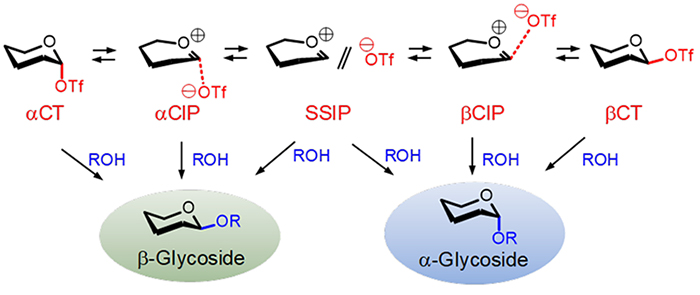
グリコシル化反応における生成物のα/β選択性は、CTや各イオンペア類から進行する求核置換反応の相対速度により決定される。よって、α/β選択性の制御法という観点からグリコシル化反応を眺めると、CTやイオンペア類の化学特性(例えば化学構造や相対エネルギー)を、アクセプターとの反応性と関連づけながら体系的に理解することが肝要であるといえる。しかしながら、図 2の平衡混合物中で、低温NMR等で実験的に検出可能な化学種は、多くの場合でαCTのみである9。これは、図 2の平衡混合物中で、αCTがアノマー効果による安定化作用により最も安定になることに主に起因する。
αCT以外の化学種についての詳細な情報を得ることは、現状では非常に難しい。このことは、αCTを低温核磁気共鳴(NMR)法等によって構造解析を行った報告がかなり存在するものの9-11、同じ共有結合性の化学種であるβCTを実際に検出した例はほとんどないことからもうかがえる12。また、より不安定で反応液中での寿命がより短いと考えられるイオンペア類の検出に関しても、ごくわずかな例を除いて報告例がない13。しかしながら、低温NMR法ではαCTのみが検出される反応系でも、その系において実際にグリコシル化反応を行うとC-1における立体化学が保持されたα-グリコシドが通常は少なからず生成し、この実験事実は図2の平衡が存在することの1つの裏付けとなっている。
このように、アクセプターとの求核置換反応に関わっている実際の化学種を検出し、その特性評価を行うことの困難さが、グリコシル化における反応機構の全容解明を困難にしている主な要因である。本事項を受けて、溶液中における図2の平衡混合物の構成要素であるCTやイオンペア類の反応性に関する情報は、間接的手法を用いることで研究されている。例えば、生成するグルコシドのα/β比へのC-1の同位体効果を調べることで、グリコシル化反応におけるイオンペア類の寄与度に関する貴重な情報を得ることが可能である14。またC-1に対して隣接基関与を引き起こす特殊なドナー分子のグリコシル化挙動から、求核置換反応におけるSɴ2性、Sɴ1性を評価した報告が存在する15。一方、直接的なイオン種の検出例としては、質量分析法や赤外分光法等によるオキサカルベニウムイオンの分析が試みられている16, 17。しかしながら、このような直接分析は高真空状態の分析装置内で行われることが多く、グリコシル化が実際に進行する溶液中のものとは異なった情報が得られる可能性がある点に注意を有する。
第一原理計算や密度汎関数理論(DFT)に根差した計算による種々の化学現象の解釈、予測は、昨今の化学者にとって一般的な研究手法の1つになっている。グリコシル化反応機構についても、特にDFT計算を用いた不安定中間体や遷移状態の計算が行われている18-24。図 2のイオンペア類は典型的な不安定化学種であり、その化学特性の評価は理論計算の得意とするところのように思われる。しかしながら、近年の理論化学的手法をもってしてもCTからのイオンペア類生成過程の正確な計算はかなり挑戦的な課題である。例えば、グリコシルトリフレートのC-1─OTf結合を伸展させることで、グリコシド結合長に関するポテンシャルエネルギー曲線をMP2法やDFT法等で計算しても、得られるエネルギープロファイルは結合距離について単調増加になってしまい、CTから直接的に生成するはずのCIPは安定構造として計算されない。また、結合解離後のオキサカルベニウムイオンとTfO−を、十分に離れた距離かつ様々な相対位置で存在させた状態で構造最適化計算を開始しても、イオンペアの構造は得られずに、初期構造におけるカチオンとアニオンの相対配置が反映されたαまたはβCTの構造が得られる。この傾向は、連続誘電体モデル等で溶媒和効果を計算に取り入れても同様である。
以上の問題を言い換えると、計算化学的な反応解析で一般的によく用いられる計算スキームを用いている限り、図 2のイオンペア類は計算上存在しないことになり、したがってイオンペア類の特性評価を計算することはできないということになる。もちろん、安定構造として得られるCTと様々なアクセプターとのSɴ2的な求核反応は、比較的容易に計算することができる。実際に、α/βCTからβ/α-グリコシドができる際のSɴ2反応の活性障壁から、実験的に求められるα/β比を予測した例はかなりの数で存在するが、定量的で正確なα/β比の予測は困難であることがほとんどである。この原因としては、採用される計算手法の精度の問題(簡便だが定量性に欠けるとされるDFT計算が採用される場合が多い)など、様々なものが考えられるが、一定の割合で進行しているはずのCIPやSSIPを起点とする種々の求核反応がそもそも考慮されていないことも主因の1つであろう。
このイオンペアが計算できないという問題を解決するための糸口は、Whitefieldらの先駆的研究によって見いだされた24。すなわち、Li+をTfO基に配位させた状態で構造最適化を行うと、オキサカルベニウムイオンとTfO−の間に穏やかな静電相互作用を有すると考えられるCIPの構造が最適化されることが報告された。この成功の原因は、C-1─OTf結合のイオン解離によってTfO基に蓄積する負電荷がLi+によって相殺されたことによると考えられる。彼らのDFT計算により、αCTのC-1─OTf共有結合の解離により4H3と呼ばれる半イス型のピラノース環立体配座を有するCIPの構造が最適化された。一方でこの計算手法は、反応系中に実際には存在しないLi+を人工的に計算系に導入しているというという問題も有している。
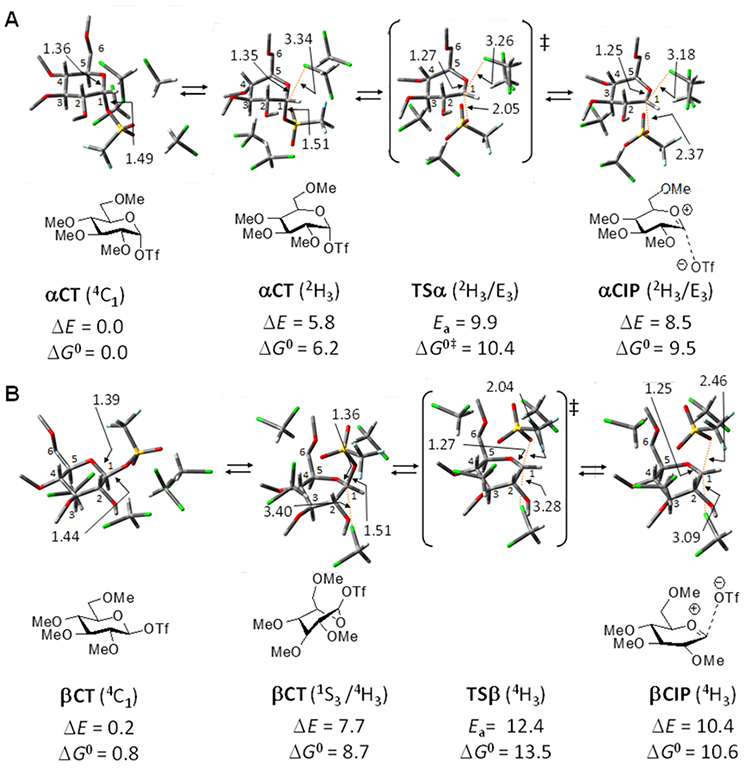
本稿の著者も、TfO基を脱離基とする一連のグリコシル化におけるイオンペア類の構造や安定性の評価を、計算化学的手法を用いて行っている。図 3Aに示したメチル基をモデル的な保護基として有する共有結合性α-グルコシルトリフレート αCTからのジクロロメタン中でのイオン解離について検討した場合、溶媒分子4分子を明示的にドナーのα/β面に配置することで、上述のようなLi+の配置を要さずに、イオンペア類が安定種として計算されることが見出された20。具体的には、MP4(SDQ)法やDFT(M06-2X)法による計算により、αCTからのαCIPの生成では、出発物のピラノース環の立体配座が最安定の4C1(イス型)から2H3(半イス型)へと変化し、その後C-1─OTfのイオン解離にともなう遷移状態TSαを経て2H3/E3立体配座を有するαCIPが生成する。このαCIPは、αCT (4C1)と比較してポテンシャルエネルギーベースで8.5 kcal/mol(Gibbsエネルギーベースでは9.5 kcal/mol)不安定であり、低温NMRでイオンペア類が検出されないこととよく対応した結果が得られている。またβ型のCTであるβCTからのCIP生成の場合では、4H3というα型の場合とは異なった立体配座を有するβCIPが生成することが示されるとともに、このCIPはαCIPと比較して1 − 1.5 kacl/mol程度不安定である(ΔE=10.4 kcal/mol, ΔG=10.6 kcal/mol)ことが判明している(図 3B)。さらにSSIPについても、様々なTfO−や溶媒分子の配置パターンのものが最適化されている。SSIP類は独立したグルコシルオキサカルベニウムイオンが好む4H3配座を取る傾向が強く、CIP類と同程度のエネルギーを有することが示されている。
同様の溶媒分子を明示的に取り扱ったイオンペア類の評価は、マンノシルドナーにも適用されている23。本研究では、上記のグルコシルドナー由来のイオンペア類が4H3などの半イス型立体配座を好むのに対し、マンノシルドナーのイオンペアはB2,5などのボート型の立体配座を好むことが報告されている。また、化学合成が非常に難しいβ-マンノシドなどの1,2-cisグリコシドの合成を可能にする保護基として報告されている4,6-ジアセタール構造の影響についても計算が行われた22。DFT(M06-2X)レベルでのマンノシルトリフレートに対する計算により、4,6-ジアセタール構造の導入によって、αCIPの他のイオンペアに対する相対的な安定性が高められることで、β-マンノシドの選択的な生成が可能になっていることが示唆されている。この考察は、C-1に対する同位体効果を用いた実験的な機構解析結果ともよく一致しており14,25、計算と実験を組みわせたさらなる検討により、グリコシル化反応機構の全容解明が行われることが期待されよう。
本稿ではグリコシル化における反応機構解明研究の現状、課題について解説した。図 1のような単純な求核置換反応であっても、その裏に潜む化学は非常に深遠であることが少しでも理解いただければ幸いである。望みの糖分子を自在に創り出す手法を化学者が手にするのは当分先のことになると思われるが、そのためには本稿で紹介したような反応制御の判断材料に足るほどの精度でグリコシル化機構を解明することが必要であろう。
著者は、計算化学に軸足を置いたグリコシド化における機構研究を今まさに展開し始めたばかりである。計算化学的研究手法は、不安定中間体や遷移状態などの構造やエネルギーを評価する際に大きな力を発揮し、まさに不安定中間体であるイオンペア類の評価を目指すうえで不可欠な手法であるといっても過言ではない。一方で、昨今の実験手法の発展も目覚ましくexchange NMR法と呼ばれる特殊なNMR分析によって、イオンペアの分析を行った例が報告されている13。今後は、実験と理論をより密に連携させた研究がこの分野で展開されることで、グリコシル化機構の解明研究がさらに発展することが期待される。



