
氏名:安藤 弘宗
岐阜大学糖鎖生命コア研究所 教授
1999年岐阜大学大学院連合農学研究科修了、同年科学技術振興事業団CREST博士研究員、2002年日本学術振興会特別研究員、2003年岐阜大学生命科学総合研究支援センター助手、2008年同大学応用生物科学部ならび京都大学物質―細胞統合システム拠点准教授、2017年岐阜大学高等研究院生命の鎖統合研究センター教授ならびに京都大学高等研究院物質―細胞統合システム拠点客員教授、2020年より現職。
シアル酸含有糖脂質の化学合成を中心に糖鎖機能の解明を加速するための分子創製とその応用について研究を進めている。
シアル酸は、カルボキシル基という酸性基を有している「糖酸」と呼ばれる糖の仲間である。生体中の糖鎖を構成する単糖の多くが五つまたは六つの炭素からなる五炭糖、六炭糖であるのに対して、シアル酸は九つの炭素からなる九炭糖であり、糖鎖の関わる生命現象において多彩かつ重要な役割を担っている。恐らく糖のコミュニティーで一目を置かれる存在であるに違いない(と筆者は信じている)。糖鎖の化学合成では、シアル酸が糖鎖に含まれるとその難易度は飛躍的に上昇する。これは、シアル酸の特殊な構造がグリコシド化反応の成功率を著しく低下させるためであり、この問題の根本解決が半世紀以上にわたり模索されてきた。最近、我々のグループはこの課題を解決する一つの手法を開発することに成功した。本稿では、その手法について概説させていただく。
化学的な(人為的な化学反応によるという意味で)グリコシド化の多くは、糖の還元末端の炭素(アノマー炭素)を電子不足の状態にして、そこに電子豊富な糖水酸基の酸素を攻撃させて、新たなC-O結合を形成させるものである。この形式の反応では、前者を糖供与体、後者を糖受容体と呼ぶ。結合形成の成否は、両者の化学的な性質によって左右される。シアル酸のグリコシド化反応では、電子不足種であるシアル酸のオキソカルベニウムイオンの性質に問題がある(図 1)。シアル酸のアノマー炭素にはカルボキシル基が結合しており、グルコースやガラクトースのように水素が結合したアノマー炭素と比べて、立体的に混みあっている。これは、水酸基による攻撃には都合が悪い。さらに、電子求引性のカルボキシル基は、オキソカルベニウムイオンを不安定化する。さらに都合の悪いことに、アノマー炭素に隣接するメチレン炭素の水素は、不安定な電子欠乏状態の解消に向けて、陽イオンとして脱離しやすく、結果、二重結合を持つ分子(2,3-エン体)へと分解する不活性化反応が競合する。これらの三つの問題がシアル酸のグリコシド化を困難にしている本質的な原因である。また、仮に水酸基がアノマー炭素を攻撃できたとしても、二つの立体異性体を与える可能性がある。何も策を講じない場合は、アノマー効果の影響により、生体糖鎖では見出されていないβグリコシドが優先して生じる傾向が強い。その為、もう一方の天然型のαグリコシドを得るためには、何らかの工夫が必要である。しかし、アノマー炭素の隣接位に水酸基を欠くシアル酸では、求核性置換基による隣接基関与を利用した確実性の高い立体制御法の利用が極めて難しい。これがシアル酸のグリコシド化を困難にするダメ押しの問題である。いわば、この四重苦を如何に克服するかが、シアル酸のαグリコシド化の課題である。少し大げさに言うと、Fisherのグリコシル化反応を化学的グリコシド化反応の端緒とすれば、シアル酸のαグリコシド化は、約120年にわたる糖化学の難題のひとつとして捉えることができる。
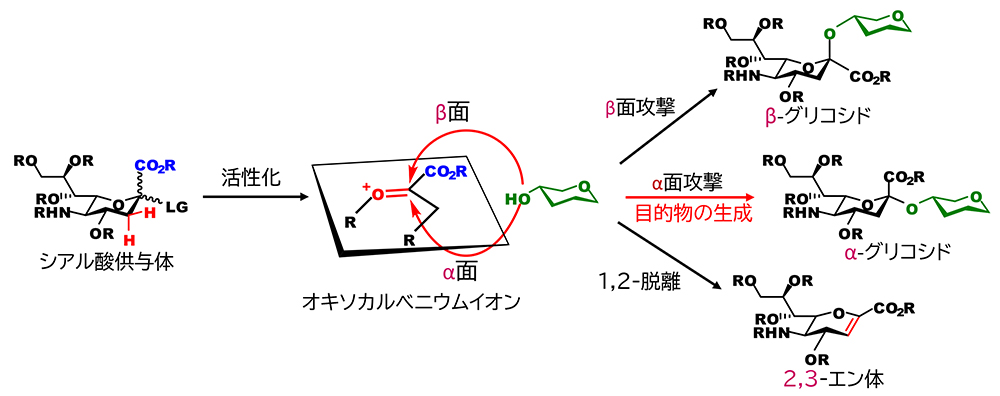
シアル酸のαグリコシド化を成功に導くためには、不安定なシアル酸のオキソカルベニウムイオン中間体の分解を抑制することと、水酸基の攻撃面をα面に限定することが重要である。これまでに効果的なα選択的シアロシド化(シアル酸のグリコシド化)のために、ニトリル溶媒効果による方法、アノメリック炭素の隣接位に補助基を導入する方法、4,5位に環状カーバメート構造を導入する方法など様々な手法が開発されてきた(詳しくは、拙文をご参照ください)1。上記の手法は、我が国の研究者が先駆けて開発したものである。いずれもオキソカルベニウムイオンのβ面を弱い結合によって遮蔽、安定化し、αグリコシドを得る方法であり、多くのシアル酸含有糖鎖の合成を導いてきた。しかしながら、反応機構上はβ体の生成の可能性があり、反応条件次第で立体選択性が変動する。実用性という観点では、立体異性体の生成は極性の酷似したシアル酸のアノマーの分離を必要とするため、完全に克服すべき課題となる。したがって、原理的に異性体が生じ得ないシアロシド化反応の開発、つまり、完全にα選択的なシアロシド化の確立の意義は大きいと考えられる。
シアル酸の特徴でもある同時にグリコシド化の妨げの要因である1位のカルボキシル基は、アノメリック炭素の立体化学がαの場合、5位アミノ基と1,4-cis配置の関係になる。我々は、これを架橋することができれば、グリコシド化が起きる際に架橋部によってβ面が完全に遮蔽され、結果的にαグリコシド体のみが得られると予想した(図 2)。
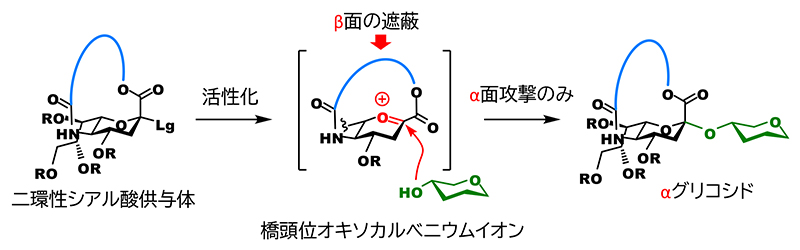
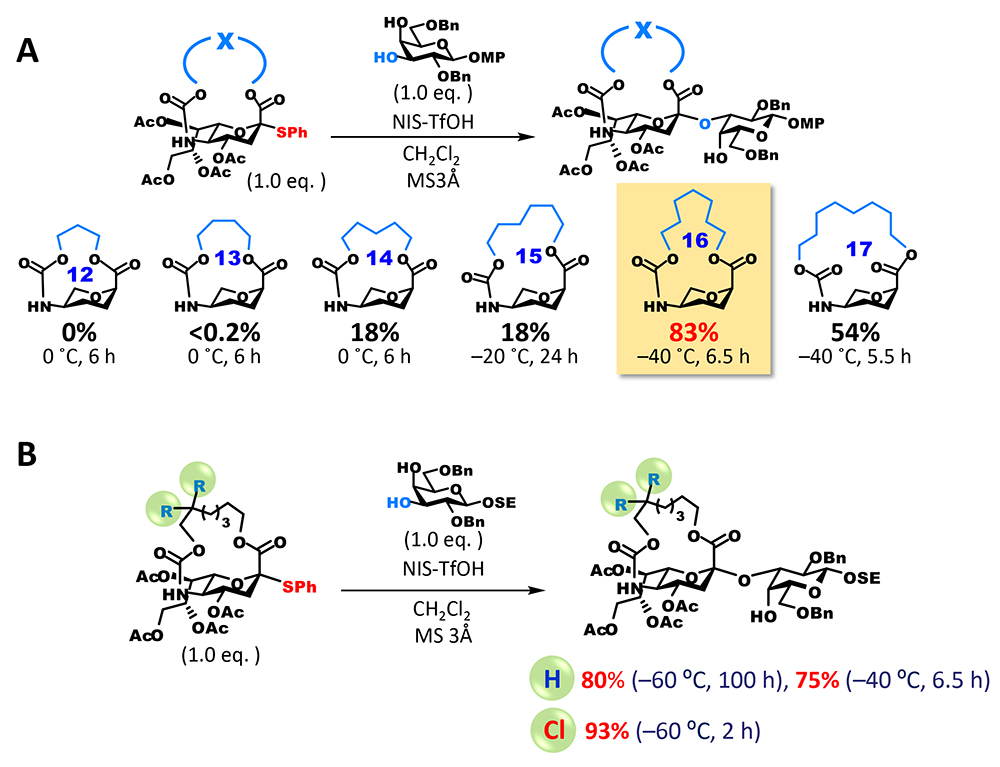
しかし、アノメリック炭素は縮環の橋頭位(二つの環の二つの結合点の内の一つ)となり、強力な環歪みにより平面構造のsp2炭素として存在するカルボカチオンの生成が圧倒的に不利である(Bredt則)。一方で、大環状構造を持つanti-Bredtのカチオンまたはオレフィンの生成は数多く報告されており3,4、これらは、架橋部の長さを整えることで橋頭位にsp2炭素を生じさせることが可能であることの示唆であると捉えた。但し、シアル酸の架橋体はアザ-オキサ環を含む二環性化合物となり、ヘテロ環が縮環した橋頭位のanti-Bredtカチオン(またはオレフィン)の構造は文献上未知である。そのため、我々は、実験的にsp2炭素形成を許容する架橋部の長さを確かめることとした(図 3A)。5位のアミノ基に炭酸エステルとして架橋部を導入した後、1位のカルボキシル基と架橋部末端の水酸基との脱水縮合により閉環するという工程で大環状部分が12員環から17員環のシアル酸供与体の候補化合物を合成した。その後、グリコシド化を検討したところ、期待通り、グリコシドとして生成した成績体は全てαグリコシドであり、16員環を有する供与体が最も高い収率を与えるという結果が得られた。興味深いことに、員数の一つの差が大きく収率に影響し、15員環ではグリコシド化が終結せずに未反応の供与体が39%回収されたのに対して、17員環では反応は完全に終結したものの、約40%は1,2-脱離によって生じたオレフィン体(2,3-エン体)が得られた。16員環は15員環に比べて環歪みが緩和され、橋頭位カチオンの生成を許容できるが、17員環と比べてアノメリック炭素の完全な平面構造がエネルギー的に不利なため、sp3炭素構築となるグリコシル化が優位になったもの推測できる。
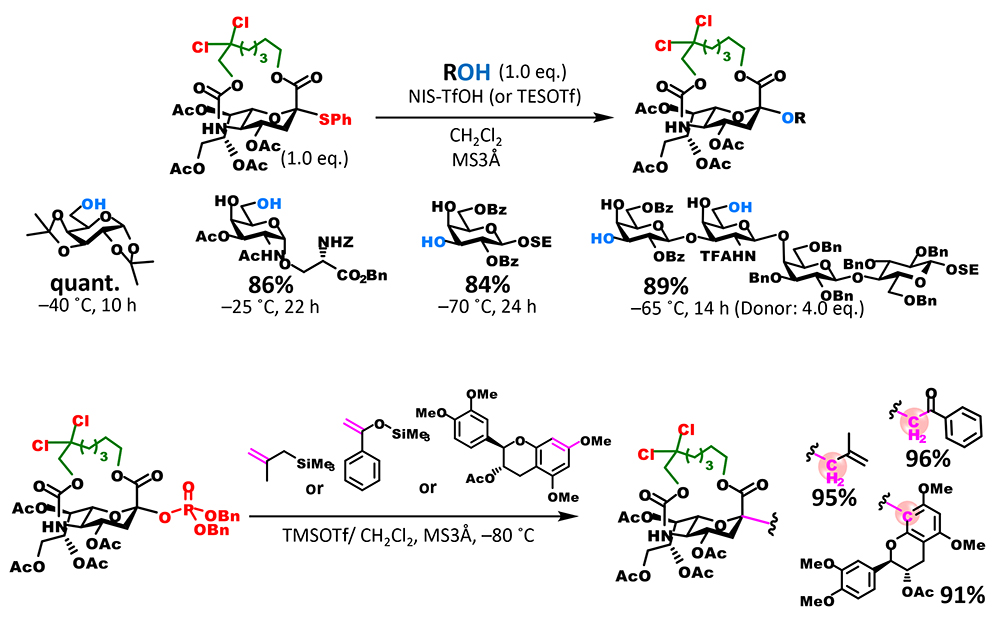
16員環体によるグリコシド化の収率は満足のいく水準であったが、反応時間が比較的長く改善の余地を認めた。また、5位カーバメート基はアルカリ加水分解等によって除去可能であるが、天然に存在するシアル酸の類縁体の合成を視野に入れると、選択的にアミノ基を遊離にできる改良が必要であると考えた。この二つの要件を満たすために、我々は、アミノ基の保護基に使用される2,2,2-trichloroethoxycarbonyl(Troc)基に着目した。Troc基は他のエステル系保護基が併存する中で選択的に除去することが可能である。また、我々はTroc基をシアル酸5位アミノ基に導入すると供与体としての反応性が飛躍的に向上することを見出していた5。この性質を大環状構造に付与するために、カーバメート部位に結合するアルキル鎖のβ位を二塩素化した修飾体を合成した。期待通り、新たな塩素二置換供与体は、無置換体を格段に上回る反応性を示し、穏和な条件かつ短時間で反応が終結し、極めて高い収率でグリコシドを与えた(図 3B)。加えて、カーバメート部位の遊離アミノ基への変換も選択的に可能であり、5位アミノ基の化学修飾が異なる種々のシアル酸類縁体の合成へと派生することができた。
基質適用範囲の検討実験で、塩素化供与体の幅広い基質適用範囲が示された(図 4)。また、脱離基をジベンジルリン酸エステル基に置き換えることで、チオグリコシド供与体の活性化条件で毀損されるオレフィンを有する基質も利用可能となった。さらに同供与体と炭素求核剤との反応によって、C-グリコシドの構築にも成功しており、基質適用範囲が更に拡張された。
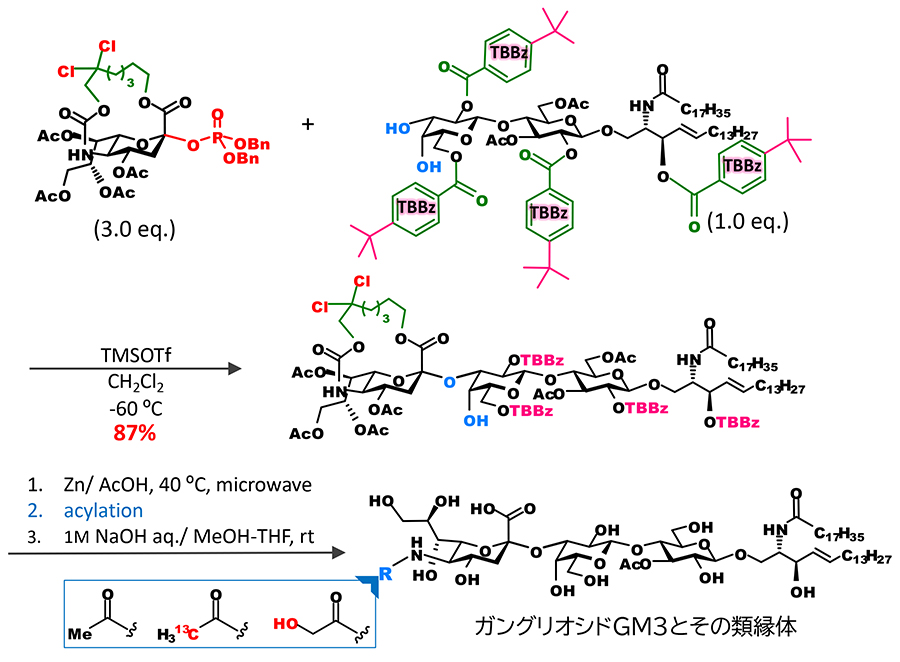
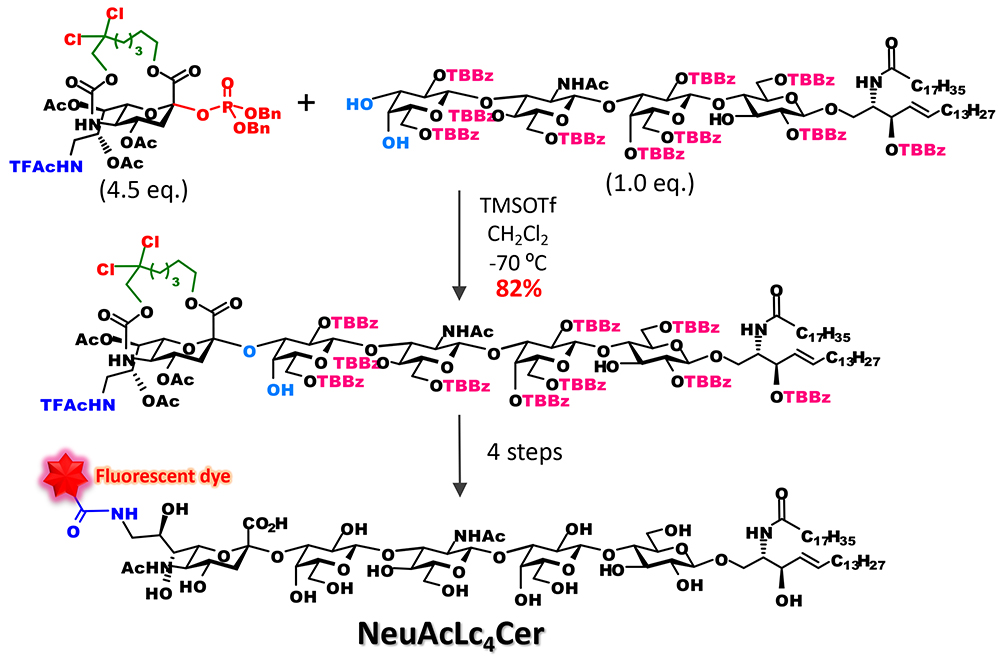
特筆すべきは、オリゴ糖受容体に対しても収率よくシアル酸を結合させることが可能となった点である。従来の合成では、立体異性体の分離を回避するために、シアル酸は単糖と結合させた二糖ブロックとして糖鎖に組み込んできた。しかし、この手法により、糖鎖末端に結合するシアル酸の導入を糖鎖伸長の最終段階で行うことが可能となった。例えば、我々は糖脂質受容体に直接シアル酸をグリコシル化することにより、ガングリオシドGM3の骨格を得て、その後に選択的な架橋部の切断、5位アミノ基のアシル化によって種々のGM3類縁体の合成が可能であることを示すことができた6(図 5)。さらにこのアプローチを糖鎖長の長いガングリオシドの蛍光プローブの合成に応用し、1分子イメージング用のラクト系ガングリオシドの蛍光プローブを開発することができた7 (図 6)。
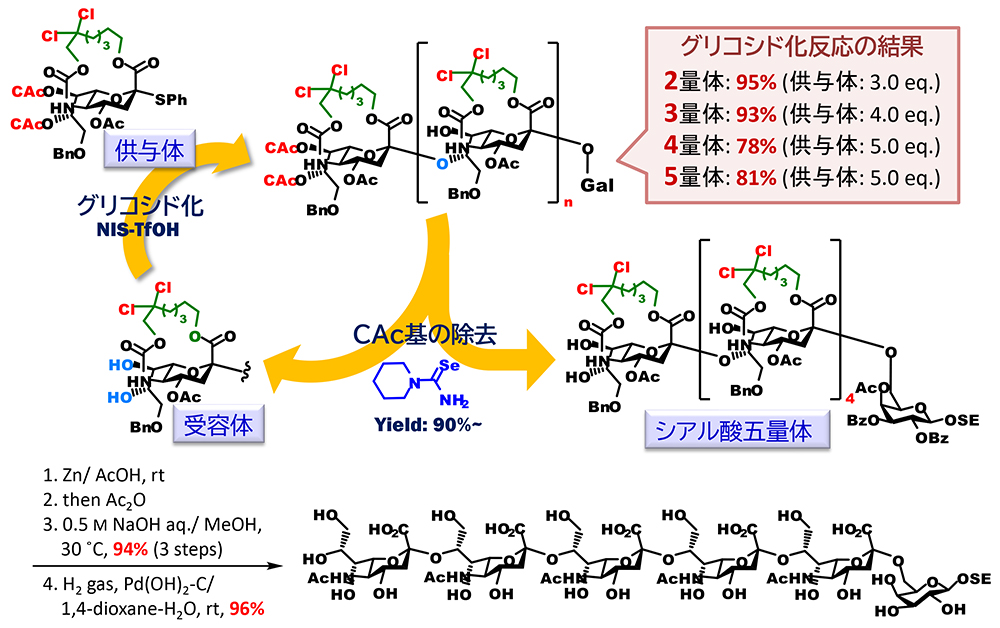
さらに我々は合成難易度の高いシアル酸の多量体の合成においても、架橋シアル酸を用いる手法が有用であることを示すことができた(図 7)。天然の糖鎖には、シアル酸のタンデム構造が存在し、糖鎖合成化学的には、シアル酸の8位水酸基でシアル酸が結合したα(2,8)オリゴシアル酸は最高難易度の構造とされている。これは、シアル酸8位水酸基の反応性が5位アミド基との水素結合によって著しく低下しているためであると推測されている。我々は、大環状化により、4位炭素と5位窒素間の結合の自由回転障壁が高くなり、8位水酸基と5位NH基間での水素結合による環形成は不利になるものと予想した。これを基に供与体と同じ縮環構造を持つ7位、8位水酸基遊離の受容体を合成し、供与体とのグルコシル化に供したところ、予想通り高収率でα(2,8)結合のシアル酸二量体を合成することに成功した。DFT計算による受容体の配座解析では、受容体は通常の椅子型配座である2C5に加えて、これが反転した5C2の椅子型配座、そして2,5Bの舟形を主たる配座としてとっていることが明らかとなった。この結果を受けて、次にシアル酸の7位、8位を選択的除去が可能なクロロアセチル基で保護した供与体を合成し、グリコシル化、クロロアセチル基の選択的除去による受容体への変換の二工程を繰り返すことでα(2,8)オリゴシアル酸の合成を試みた。その結果、五量体までの伸長に成功し、得られた五量体は、架橋部の選択的切断、N-アセチル化、鹸化、ベンジル基の除去を経て、無保護体へと変換した。
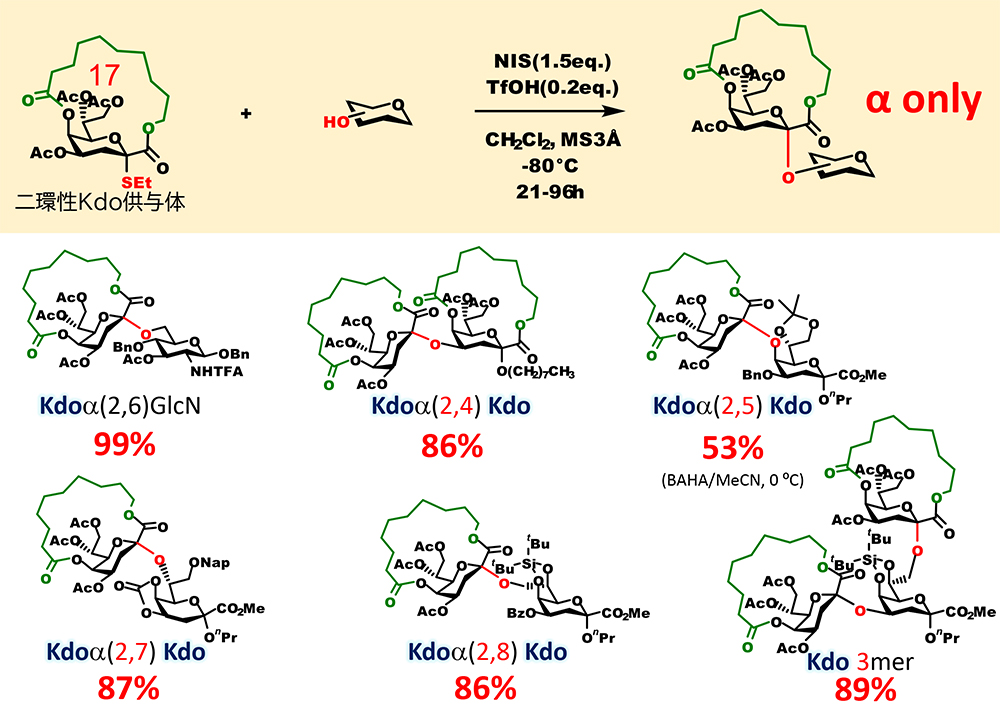
また、我々は最近、上述の二環性大環状構造によるグリコシド化の立体制御をシアル酸と構造が類似した2-keto-3-deoxyoctulosonic acid(Kdo)のαグリコシド化に応用することが8,9(図 8)。Kdoは細菌の外膜に存在するリポ多糖や莢膜多糖の構成糖残基であり、Kdo含有糖鎖は細菌の感染や宿主体内での免疫回避に関与している。Kdoのαグリコシド化の立体選択性を恒常的に得るための信頼性の高い方法が開発されてきたが、シアル酸と同様に完全にβ異性体の生成を排除する仕組みは存在していなかった。我々は、大環状化したKdoの構造検討により17員環の大環状構造を持ち、脱離基としてエチルスルフェニル基を有する供与体が最適であることを見出した。この二環性Kdo供与体を用いることで細菌の糖鎖中のKdoタンデム構造の立体選択的合成に成功した。
以上述べたように,我々は橋頭位オキソカルベニウムイオンを求電子種として用いることで、これまでに不可能であったシアル酸の完全立体選択的αグリコシド化を実現した。これまで最も難易度が高いとされていたシアル酸グリコシド化反応は、現時点で(立体異性体が生じないという意味で)最も簡便なグリコシド化反応となったと言える。このことは、シアロ糖鎖合成の合理化、シアル酸と他の機能性分子との複合化を簡便化するものと期待できる。また、シアル酸のグリコシル化の立体制御の問題は、糖鎖合成の自動化の隘路ともなっているため、本反応の応用によるさらなる発展が望める。また、同様に橋頭位オキソカルベニウムイオンを反応種とするKdoの完全な選択的αグリコシド化を実現した。糖鎖の生物学的機能の解明を推進するため、またその機能を医薬等へと応用するためにも糖鎖合成は不可欠であり、その技術基盤を進化させ強化する努力を継続することが肝要である。本稿で紹介したグリコシド化法も改良の余地を残しており、実用性を充足させる手法へと発展させていきたい。



