
氏名:野上 敏材
鳥取大学工学部化学バイオ系学科 教授
1999年京都大学工学部工業化学科卒業。2004年京都大学大学院工学研究科博士後期課程修了博士(工学)、スイス連邦工科大学チューリヒ校博士研究員。2005年京都大学大学院工学研究科助手、2007年同助教、2011年同講師。2012年鳥取大学准教授、2019年同教授。
京都大学工学部工業化学科を卒業後、同大学院に進学し、吉田潤一先生の下で学位を取得。有機合成化学を専門とする。スイスでの11か月間の博士研究員を経て、京都大学大学院工学研究科合成・生物化学専攻・吉田潤一教授研究室の助手に着任し、有機電解合成に関する研究をスタートさせる。電気化学的手法を用いてチオグリコシドをグリコシルトリフラート中間体に変換できることを見出し、オリゴ糖の液相電解自動合成へと研究を展開した。
オリゴ糖を人工的に合成する手法は酵素法と化学法とに大別され、それぞれに様々な合成手法が存在している。酵素法は糖転移酵素あるいは糖加水分解酵素を用いてオリゴ糖を合成する手法であり、オリゴ糖の水酸基に保護基を導入せずに合成出来る点にメリットがある。一方、化学法は有機合成化学的手法に基づいてオリゴ糖を合成するため、水酸基の保護・脱保護の工程が必須となるが、天然・非天然のオリゴ糖を自在に合成出来る点にメリットがある。オリゴ糖の化学法による合成のために様々なビルディングブロックがこれまでに開発され、同じビルディングブロックについても何種類もの活性化法が存在している。本項では取り扱い容易なビルディングブロックとして幅広く利用されているチオグリコシドを、電気化学的に活性化してグリコシド結合を形成する電解グリコシル化反応について紹介する。
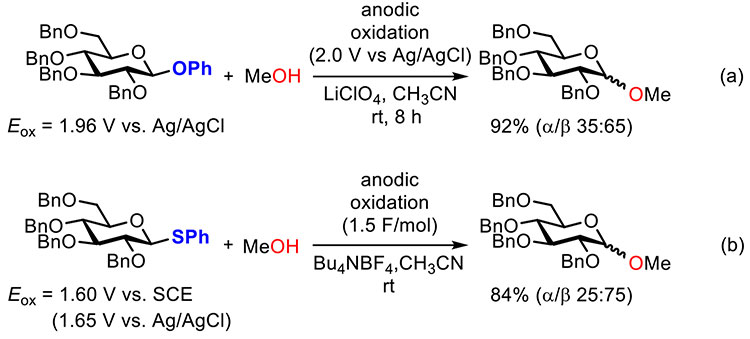
電気化学的にビルディングブロックを活性化する電解グリコシル化反応は野依、栗本らによって初めて報告された(図 1a)1。グルコースの糖アノマー炭素上に電子豊富なフェニルオキシ(PhO)基を有するフェニルグリコシドをビルディングブロックに用いて、電解酸化条件(銀-塩化銀電極基準で2.0 Vの定電位条件)での活性化に伴うグリコシド結合の形成を実現している。その後、より酸化電位(Eox)が低く活性化の容易なフェニルチオ(PhS)基を糖アノマー炭素上に有するチオグリコシドを用いた定電流条件下(電気量は1.5 F/mol、電流値は文献記載なし)での電解グリコシル化反応も報告されている(図 1b)2,3。このように電解グリコシル化反応は化学的安定性の高く、取り扱いも容易なチオグリコシドを室温・中性条件で活性化できる極めて有用な方法であるにも関わらず、筆者が研究に着手するまで3糖以上のオリゴ糖の合成にはほとんど利用されていなかった4。
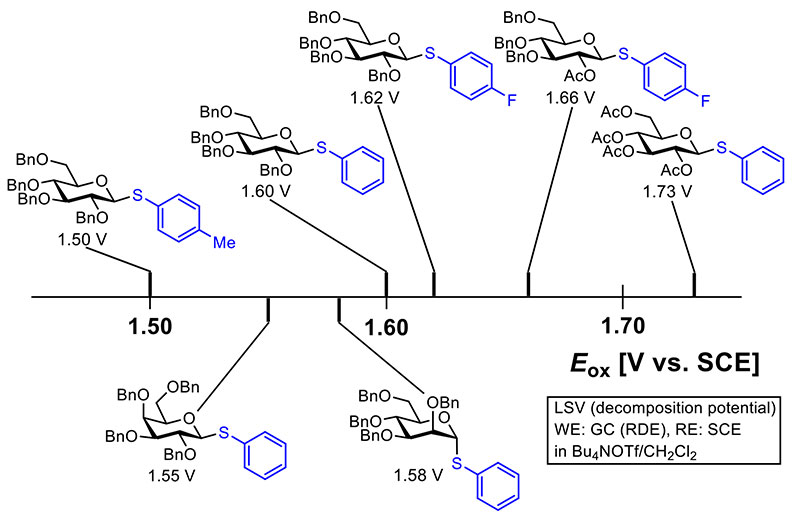
電解グリコシル化反応に限らず、電気化学的手法に基づく有機化学反応の特徴は、反応に用いる基質の反応性を酸化電位あるいは還元電位で定量的に評価できる点にある5。酸化電位測定に用いる基準電極が異なる場合は、電位を補正して直接の比較も可能である。ただし、溶媒や支持電解質といった測定条件による影響も無視出来ないため、可能な限り同一の測定条件で得られた酸化電位を比較するのが望ましい。参考までに筆者らが回転ディスク電極(RDE)を用いて測定したチオグリコシドの酸化電位を図 2に示す。チオグリコシドの酸化電位は硫黄原子が一電子酸化を受けるのに必要な電極電位であるため、硫黄原子上置換基の電子的影響を強く受ける。従って、同じグルコースからなるチオグリコシドでもフェニル基の場合(1.60 V)に比べて、電子供与性の置換基(Tol基: トリル基=パラメチルフェニル基)が酸化電位を低下させ(1.50 V)、電子求引性の置換基(4-FC6H4基:パラフルオロフェニル基)が酸化電位を上昇させる(1.62V)。加えて、糖水酸基の保護基の有無やその種類、糖の種類によっても酸化電位は変化する。すなわち、アセチル(Ac)基といった電子求引性の保護基が酸化電位を上昇させ(1.66 V)、数が多いほどその効果は顕著になる(1.73 V)。また、同じ置換基・保護基を有していても、ガラクトース(1.55 V)やマンノース(1.58 V)の方がグルコース(1.60 V)よりも酸化電位は低い。なお、酸化電位は計算科学的にも予測可能であり、密度半関数(B3LYP)により求めたチオグリコシドのHOMOエネルギー差を比較することによって、チオグリコシド間の酸化電位差を大まかに見積もる事ができる6,7。
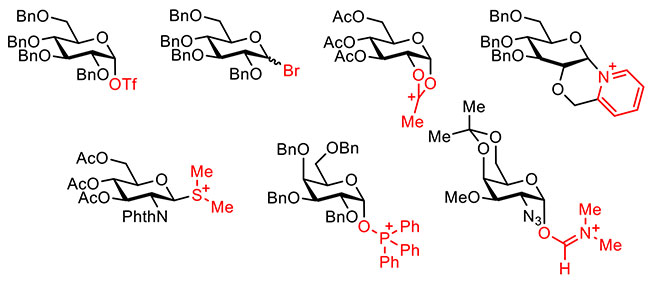
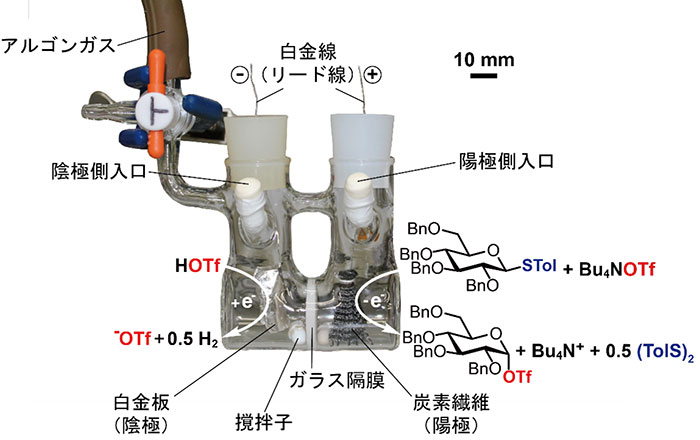
化学法によるグリコシル化反応の進歩に伴い、脱離基にハロゲン原子のアニオンを有するグリコシルハライド中間体や脱離基にトリフラートアニオン(TfO-)を有するグリコシルトリフラート中間体をはじめとするさまざまな反応中間体が観測されるようになった(図 3)8。電解グリコシル化反応はグリコシルカチオン中間体の発生が提案されていたが、実際にどのような中間体を経由して反応が進行しているか確かめられてはいなかった。そこで筆者らは共同研究者と協力して、図 4に示すような電解セルを用いてテトラブチルアンモニウムトリフラート(Bu4NOTf)の存在下、低温条件でチオグリコシドの電解酸化を行い、グリコシルトリフラート中間体9,10,11,12やグリコシルジオキサレニウムイオン中間体13を発生・蓄積した。これらの中間体は電解反応後の陽極溶液を、低温条件下でNMR測定することで確認されている。また、生じた中間体が糖水酸基とグリコシド結合を形成することも分かっている。
これまでにもオリゴ糖を樹脂ビーズや多孔性金属などの担体に固定して糖鎖伸長を行う、オリゴ糖の固相自動合成が報告されていた14。しかし、固相合成では得られるオリゴ糖の量が少なく、合成に大過剰の試薬を必要とするため、液相合成(基質や試薬が溶媒に溶けた状態で行う均一系での反応に基づく合成)の自動化が望まれていた(表 1)。電気化学的にグリコシル化反応中間体が発生・蓄積できることがいったい何の役に立つのか、筆者が研究を始めた当初はぼんやりとしていた。しかし定量的に中間体が発生し、その中間体がグリコシド結合形成に有用であることを踏まえ、オリゴ糖の液相電解自動合成へと展開できたので、以下で紹介する。
| 比較項目 | 固相合成 | 液相合成 | 反応追跡 | 大過剰必要 | 理論量~小過剰 | 試薬量 | 担体から切り出した上で行う | 溶液のまま分析が可能 | 反応の繰り返し | 可能 | 不可能 | 洗浄操作 | 容易(必要) | 困難(不必要) | スケールアップ | 困難 | 容易 |
|---|
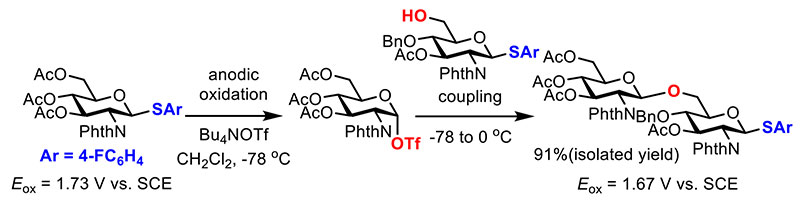
電解グリコシル化反応に基づくオリゴ糖の液相電解自動合成において最も重要な部分は、蓄積したグリコシルトリフラート中間体に対するビルディングブロックの反応である(図 5)15。ここで用いるビルディングブロックはグリコシルトリフラート中間体とグリコシド結合を形成する無保護の水酸基と電気化学的に活性化可能なアノマー位の脱離基の両方を備えている必要がある。想定される副反応としてはグリコシルトリフラート中間体と脱離基との反応であるが、脱離基である硫黄原子上に電子求引性の置換基(パラフルオロフェニル基)を導入することで、目的の水酸基とのグリコシル化反応が選択的に進行した。また、得られた2糖の酸化電位(1.67 V)が単糖の酸化電位(1.73 V)よりも0.06 V低いため、対応する2糖以上の長鎖オリゴ糖の場合も対応するグリコシルトリフラート中間体が電気化学的に発生できることが示唆された。
筆者らは通電によるグリコシルトリフラート中間体の発生と単糖ビルディングブロックの添加・昇温によるカップリングという操作を自動で行うために、既存の装置を組み合わせて自動合成装置を構築した(図 6)。当初は極低温反応装置やシリンジポンプ、直流電源といった装置をシーケンサーで制御していたが、のちにパソコンで制御できるよう改良した。最新の自動合成装置ではすべての装置が躯体に収納されており、各装置のパラメータが集中管理できるようになっている。

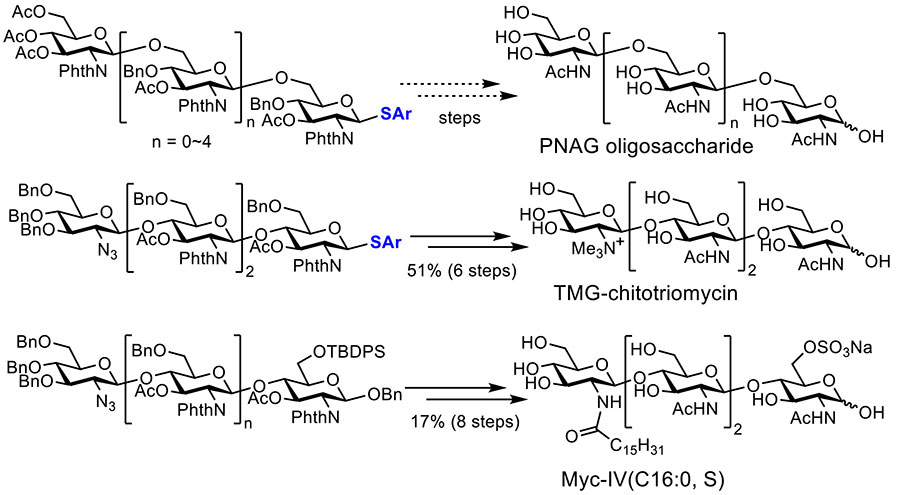
これまでに液相電解自動合成装置を用いて合成したオリゴ糖前駆体として、β-1,6-ポリN-アセチルグルコサミン(PNAG)オリゴ糖6糖前駆体15、TMG-キトトリオマイシン4糖の前駆体6,16、Myc-IV(C16:0, S) 4糖の前駆体17、環状βグルカン12糖の繰り返し構造6糖の前駆体18などが挙げられる(図 7)。一部の前駆体については脱保護により、天然物であるオリゴ糖の構造にまで誘導されている。今後、ビルディングブロックの置換基・保護基や電解グリコシル化反応条件の最適化により、収率の向上や合成可能なオリゴ糖のバリエーションが増えると期待している。
本稿では電解グリコシル化反応について、先駆的研究と筆者らの研究を中心に紹介させて頂いた。電解グリコシル化反応はあくまで化学法の一種という位置づけであり、電解グリコシル化反応でなければ実現できないグリコシド結合はないかもしれない。しかし、今回紹介した合成装置は分析装置や精製装置と組み合わせることで、機械学習による反応条件の最適化やオリゴ糖の合成から単離精製までの自動化が実現する可能性が高い。また、オリゴ糖合成以外の有用物質生産にも応用可能と考えており、電解グリコシル化反応によるオリゴ糖合成をモデル合成として、電気化学的手法に基づく液相自動合成の可能性も示せたと考えている。



