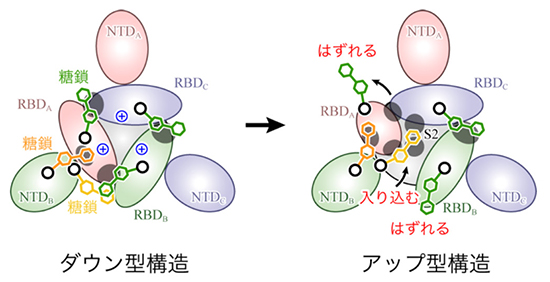
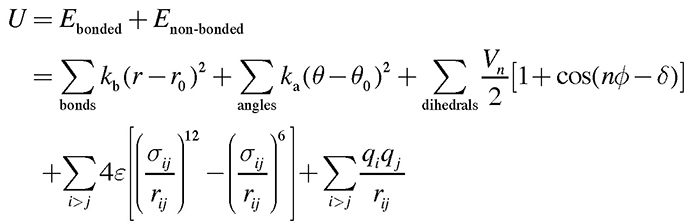スパイクタンパク質の構造変化の分子機構を明らかにするために、筆者らはスパイクタンパク質に対して分子動力学(Molecular Dynamics; MD)シミュレーションを行った。MDシミュレーションとは、計算機の中に仮想的に分子システムを構築し、各原子に対してニュートンの運動方程式 F = ma を適用することで、分子の構造や動きを予測する方法である。力Fは系のポテンシャルエネルギーの原子位置に関する1階微分から計算される。ポテンシャルエネルギーは力場とも呼ばれ、一般的に以下のような式が用いられる。
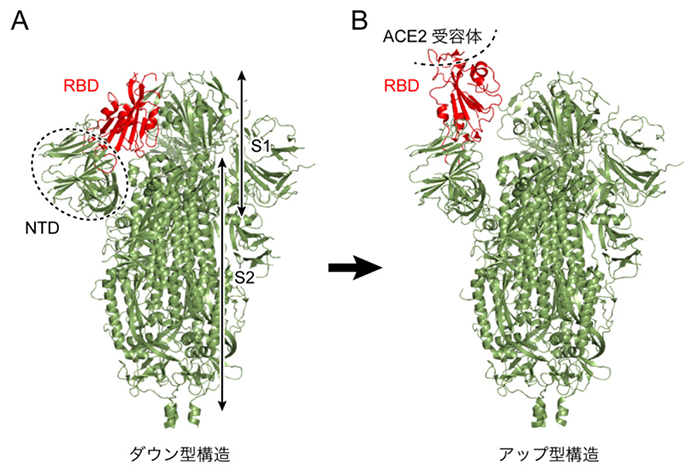
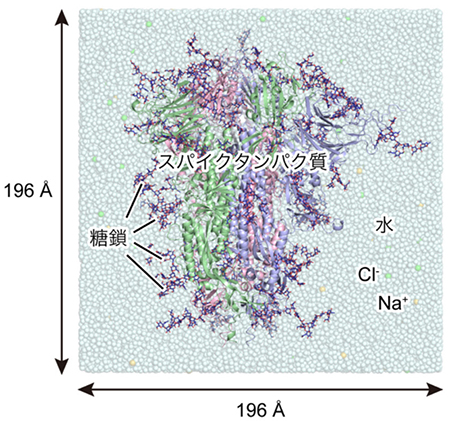
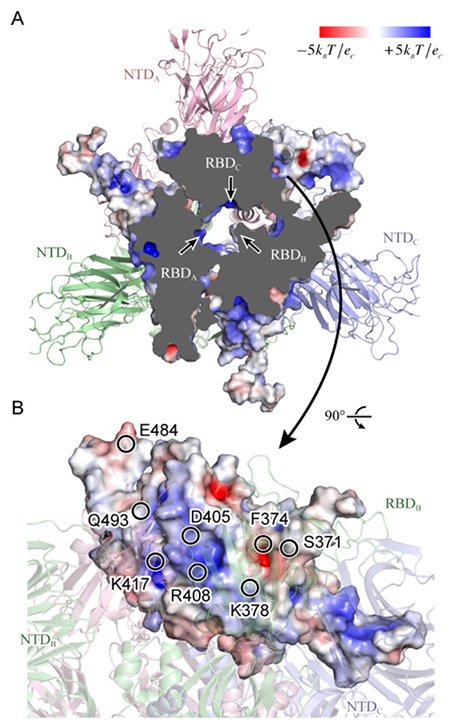
本研究で用いた分子システムを図 2に示す。スパイクタンパク質は本来膜タンパク質であるが、本研究では水溶性部分のみを切り出し、150 mM NaCl溶液中に浸した。ダウン型とアップ型の2つの系を用意し、実験データに基づいて66箇所のアミノ酸に糖鎖を修飾させている。系全体の総原子数は水分子も含めると約76万個であり、これは近年の典型的な計算規模である20〜30万原子系と比べても巨大である。
Walls, A. C.; Park, Y. J.; Tortorici, M. A.; Wall, A.; McGuire, A. T.; Veesler, D., Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell2020, 183, 1735.
Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; Wang, X., Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature2020, 581, 215-220.
Wrobel, A. G.; Benton, D. J.; Xu, P.; Roustan, C.; Martin, S. R.; Rosenthal, P. B.; Skehel, J. J.; Gamblin, S. J., SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat Struct Mol Biol2020, 27, 763-767.
Zhou, T.; Tsybovsky, Y.; Gorman, J.; Rapp, M.; Cerutti, G.; Chuang, G. Y.; Katsamba, P. S.; Sampson, J. M.; Schon, A.; Bimela, J.; Boyington, J. C.; Nazzari, A.; Olia, A. S.; Shi, W.; Sastry, M.; Stephens, T.; Stuckey, J.; Teng, I. T.; Wang, P.; Wang, S.; Zhang, B.; Friesner, R. A.; Ho, D. D.; Mascola, J. R.; Shapiro, L.; Kwong, P. D., Cryo-EM structures of SARS-CoV-2 spike without and with ACE2 reveal a pH-dependent switch to mediate endosomal positioning of receptor-binding domains. Cell Host Microbe2020, 28, 867-879 e5.
Watanabe, Y.; Allen, J. D.; Wrapp, D.; McLellan, J. S.; Crispin, M., Site-specific glycan analysis of the SARS-CoV-2 spike. Science2020, 369, 330-333.
Watanabe, Y.; Bowden, T. A.; Wilson, I. A.; Crispin, M., Exploitation of glycosylation in enveloped virus pathobiology. Biochim Biophys Acta Gen Subj2019, 1863, 1480-1497.
Jo, S.; Kim, T.; Iyer, V. G.; Im, W., CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem2008, 29, 1859-65.
Park, S. J.; Lee, J.; Qi, Y.; Kern, N. R.; Lee, H. S.; Jo, S.; Joung, I.; Joo, K.; Lee, J.; Im, W., CHARMM-GUI Glycan Modeler for modeling and simulation of carbohydrates and glycoconjugates. Glycobiology2019, 29, 320-331.
Jung, J.; Mori, T.; Kobayashi, C.; Matsunaga, Y.; Yoda, T.; Feig, M.; Sugita, Y., GENESIS: A hybrid-parallel and multi-scale molecular dynamics simulator with enhanced sampling algorithms for biomolecular and cellular simulations. Wiley Interdiscip Rev Comput Mol Sci2015, 5, 310-323.
Kobayashi, C.; Jung, J.; Matsunaga, Y.; Mori, T.; Ando, T.; Tamura, K.; Kamiya, M.; Sugita, Y., GENESIS 1.1: a hybrid-parallel molecular dynamics simulator with enhanced sampling algorithms on multiple computational platforms. J Comput Chem2017, 38, 2193-2206.
Sugita, Y.; Okamoto, Y., Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett1999, 314, 141-151.
Mori, T.; Kulik, M.; Miyashita, O.; Jung, J.; Tama, F.; Sugita, Y., Acceleration of cryo-EM flexible fitting for large biomolecular systems by efficient space partitioning. Structure2019, 27, 161−174.
Yagi, K.; Yamada, K.; Kobayashi, C.; Sugita, Y., Anharmonic vibrational analysis of biomolecules and solvated molecules using hybrid QM/MM computations. J Chem Theory Comput2019, 15, 1924-1938.
Oshima, H.; Re, S.; Sugita, Y., Prediction of protein-ligand binding pose and affinity using the gREST+FEP method. J Chem Inf Model2020, 60, 5382-5394.
Mori, T.; Jung, J.; Kobayashi, C.; Dokainish, H. M.; Re, S.; Sugita, Y., Elucidation of interactions regulating conformational stability and dynamics of SARS-CoV-2 S-protein. Biophys J2021, 120, 1060-1071.
Baker, N. A.; Sept, D.; Joseph, S.; Holst, M. J.; McCammon, J. A., Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci U S A2001, 98, 10037-41.
Mansbach, R. A.; Chakraborty, S.; Nguyen, K.; Montefiori, D. C.; Korber, B.; Gnanakaran, S., The SARS-CoV-2 Spike variant D614G favors an open conformational state. Sci Adv2021, 7.
Singh, J.; Samal, J.; Kumar, V.; Sharma, J.; Agrawal, U.; Ehtesham, N. Z.; Sundar, D.; Rahman, S. A.; Hira, S.; Hasnain, S. E., Structure-function analyses of new SARS-CoV-2 variants B.1.1.7, B.1.351 and B.1.1.28.1: Clinical, diagnostic, therapeutic and public health implications. Viruses2021, 13.
Acharya, A.; Lynch, D. L.; Pavlova, A.; Pang, Y. T.; Gumbart, J. C., ACE2 glycans preferentially interact with SARS-CoV-2 over SARS-CoV. Chem Commun2021, 57, 5949-5952.