氏名:木下 タロウ
大阪大学微生物病研究所・免疫不全疾患研究分野
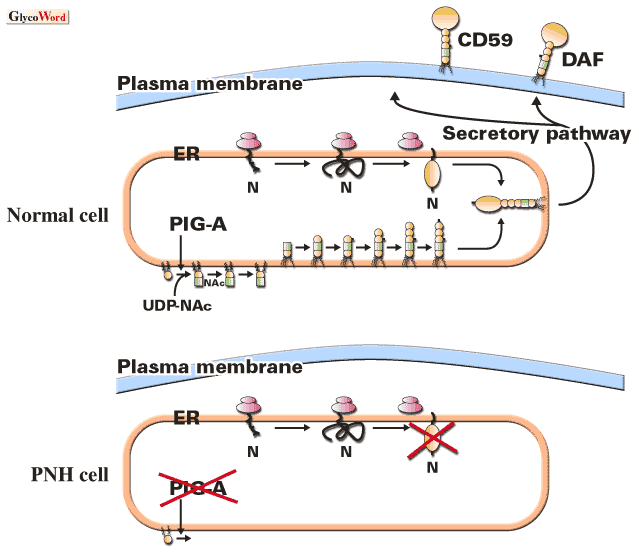
発作性夜間血色素尿症(PNH)は後天性の血液疾患で、多くは成年期に発症し、数万人に1人の割合で患者が存在する。主たる臨床症状は血管内溶血である。慢性的な溶血に加えて、感染などに伴って溶血発作が起こり、また、睡眠時に溶血が亢進することから病名が由来している。患者の血中には補体に弱い異常赤血球の集団が出現していて、補体によって血管内溶血が起こる。正常な赤血球は、活性化した補体の成分を失活させる補体制御因子であるDAFとCD59の2つのGPIアンカー型タンパク質を表面に発現しており、補体による傷害から保護されている。PNH患者の異常赤血球はGPIアンカー生合成の欠損によりDAFとCD59の両方を欠損しているため、補体の作用に著しく弱い。GPIアンカーの生合成欠損を起こす異常は多能性造血幹細胞に存在し、その細胞由来の赤血球や好中球に異常が存在する。
GPIアンカー型タンパク質は、小胞体で別々に合成されたタンパク質部分とGPIアンカー部分が合体してできあがる。GPIアンカーの生合成は、ホスファチジルイノシトール(PI)にNムアセチルグルコサミン(GlcNAc)がUDP-N-アセチルグルコサミンから転移する反応で始まり、すくなくとも9つのステップを経て完成する。この生合成には20数個の遺伝子が関与していると予想され、現在までに20遺伝子がクローニングされている。PNH患者の異常血液細胞では、この生合成経路の第1ステップの反応が起こっていない。すなわち、異常血液細胞ではPI:UDP-GlcNAc N-アセチルグルコサミン転移酵素 (GPI-GnT)が欠損している。GPI-GnTは少なくとも5つのタンパク質からなる複合体で、PNHの異常細胞では、これらの5つの遺伝子のうちのPIG-Aに体細胞突然変異が起こっている。
PIG-A遺伝子は、x染色体のxp22.1の領域に存在している。このことは、男女を問わずひとつの体細胞突然変異でGPIアンカー欠損が起こり得ることを示している。これに比べ、常染色体にある遺伝子は両アレルに突然変異が起こる必要がある。しかし、1つの細胞においてある遺伝子の両アレルに体細胞突然変異が起こる確率は非常に低いであろう。現在までに、150人以上のPNH患者の異常細胞が分子レベルで解析され報告されているが、すべてPIG-Aの変異によってGPIアンカー欠損が起こっている。これは、PIG-A以外のGPI生合成遺伝子がおそらくすべて常染色体上にある事を示唆している。実際、他の遺伝子は様々な常染色体に散らばって存在している。
PIG-Aの体細胞突然変異によってGPIアンカー型タンパク質の発現が欠損した多能性造血幹細胞ができることが、PNH発症の第1要因であるが、もし、その異常幹細胞が他の多くの正常幹細胞と同じ効率で血液細胞を供給するのであれば、臨床症状は現れないであろう。実際には、患者血球のかなりの割合を異常細胞が占めている。未熟な造血細胞に特徴的なCD34を発現している細胞集団で、すでにGPI欠損細胞が高い割合を占めていることが示されており、異常細胞クローンが分化の早い段階で拡大していると考えられる。この異常クローンの拡大が、PNH発症の第2の要因である。どのようなメカニズムで異常幹細胞が拡大するのか、2つの仮説がある。ひとつは、病的な環境が骨髄内に起こりその環境下では、正常幹細胞がより障害を受けやすく、GPIアンカー欠損幹細胞が選択的に残ってくるとする説である。ふたつ目は、PIG-Aが変異した異常幹細胞に、さらに別の遺伝子変異が重なり、それらの変異の総合で拡大能を獲得するとする説である。異常クローンが拡大するメカニズムが今後解明されなければならない。