
氏名:佐藤 一樹
東京理科大学薬学部生命創薬科学科嘱託特別講師
2013年に東京大学大学院新領域創成科学研究科で和田猛先生の指導の元、博士(生命科学)の学位を取得。東レ株式会社の研究所で5年間勤務後、2018年に東京理科大学薬学部生命創薬科学科和田猛研究室の助教に就任し、2023年に嘱託特別講師に昇任。学生時代からホスフェート構造を有する糖鎖合成の研究に従事し、東京理科大学に赴任後は核酸、ペプチドの合成研究も進めている。
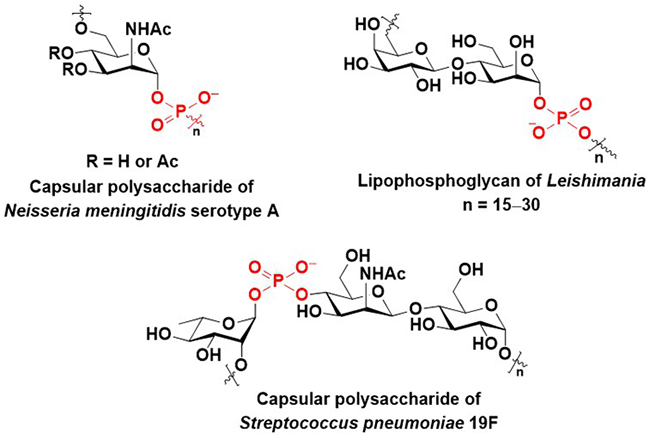
グリコシルホスフェートは糖のアノマー位にリン酸を有する誘導体である。これらは、一部の肺炎球菌1や髄膜炎菌2の莢膜多糖に繰り返し構造として見られる(Figure 1)。莢膜多糖は抗原として認識される一方で3、より抗原性の高い部分を覆い隠すことにより、宿主の免疫応答を回避する役割を担う4,5。さらに、Leishmania原虫のLipophosphoglycan(LPG)にも、グリコシルホスフェートの繰り返し構造が見られる(Figure 1)6。LPGはLeishmaniaの生存に必須のものであり、抗原として認識される部位でもある6。グリコシルホスフェートを含む生体分子を効率的に合成することができれば、こうした病原体に対するワクチンの開発や、上記多糖類の生物学的な機能の解明を通じて、治療薬の開発につながる可能性がある。
筆者らの研究グループは、このグリコシルホスフェート構造を効率的に合成するにあたり、種々のホスホン酸誘導体を用いた手法を開発してきた。その詳細について紹介する。
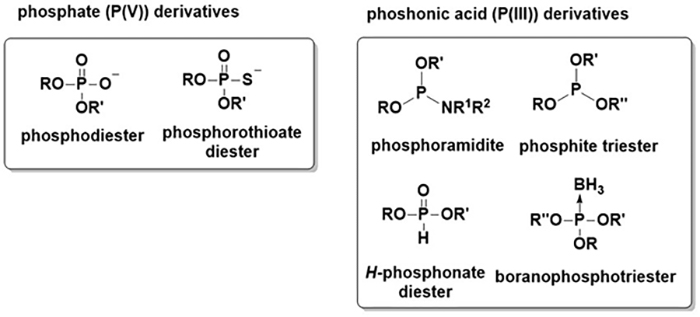
リンはヒトの体の構成元素の1%を占めるが、大半は酸化数が+5のリン酸の状態で存在している7。一方、リン原子の酸化数が+3の誘導体はホスホン酸と呼ばれる。代表的なホスホン酸誘導体としては、ホスホロアミダイト、ホスファイト、H-ホスホネートなどが挙げられる。混同しやすいが、H-ホスホネートも酸化数+3のホスホン酸の誘導体である。筆者が研究対象の中心としているボラノホスフェートは、ホスファイトのリン原子上の非共有電子対がボラノ基に配位結合している構造をしており、これもリン原子の酸化数が+3のホスホン酸の誘導体である(Figure 2)。
これまでグリコシルホスフェートの誘導体は、H-ホスホネート法と呼ばれる方法で行われてきた8。これは、グリコシルH-ホスホネートモノエステル体と、糖水酸基を、ピバロイルクロライドなどの脱水縮合剤を用いて縮合し、糖鎖間のH-ホスホネートジエステル結合を形成する。このH-ホスホネートジエステル結合は、特に塩基性条件下、水などの求核種が存在する状態では分解されやすいため、より安定なホスホジエステル体へと酸化する。その後、糖水酸基の保護基を脱保護し、再びH-ホスホネートモノエステル体との縮合反応を行うことで、鎖長伸長が可能である。この手法は、比較的鎖長が短いグリコシルホスフェート誘導体の合成時には効率的に縮合反応が進行すること、さらにはα体が優先的に得られやすいという特徴を有する8。特に後者に関し、天然に存在するグリコシルホスフェート誘導体は、アノマー効果の影響からかα体が多く存在するため、本手法は生体分子の合成に向いており、盛んに用いられてきた側面がある。
しかし、この手法も長鎖のオリゴマー合成時には課題がある。前述の通り、縮合反応で生じたH-ホスホネートジエステル体を酸化してホスホジエステル体へと誘導することが一般的だが、その反応はピリジン中、ヨウ素と水による酸化が用いられる。しかし、この反応条件ではH-ホスホネートジエステル体の加水分解との競争反応になり、このことで糖鎖の切断が起こりやすい。特にこの反応は鎖長が長くなるほど顕著である9,10。さらに、たとえホスホジエステル体へと誘導できたとしても、糖鎖間結合がアニオンを有することになり、次の縮合反応時に縮合剤と反応して系が複雑化することが懸念される。以上の理由からかH-ホスホネート法を用いたグリコシルホスフェート繰り返し構造の合成は、鎖長を制御した系ではホスホジエステル結合を4か所有するものが最長である11。
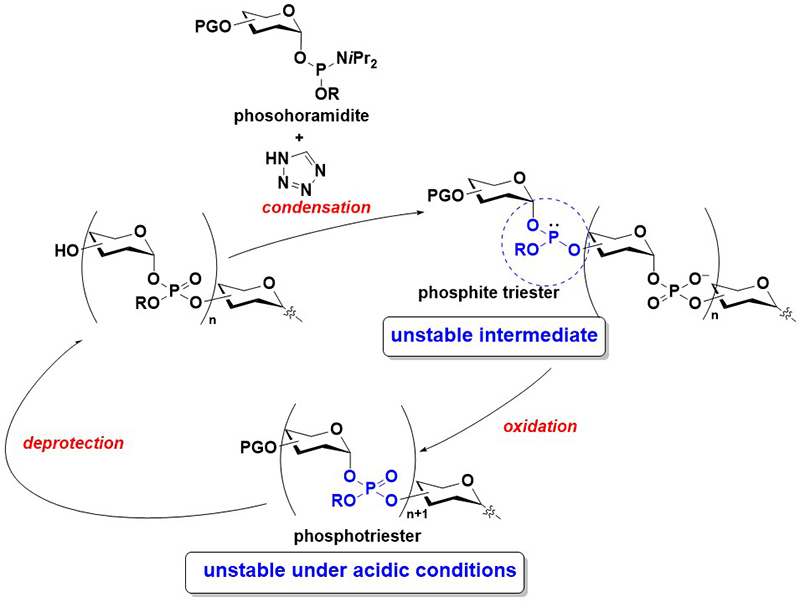
一方、ホスホジエステル結合の形成法として、核酸化学の分野ではホスホロアミダイト法が汎用される12。この手法では、同じくホスホン酸の誘導体であるホスホロアミダイトをモノマーとし、テトラゾールなどの酸性活性化剤存在下、水酸基と反応させることでホスファイトトリエステルを得たのち、これを酸化することで電気的に中性なホスホトリエステル体へと誘導する。その後、水酸基の脱保護を行い、さらにホスホロアミダイトモノマーと反応させることで鎖長伸長を行う。ホスホトリエステル体は合成の最終段階で脱保護し、ホスホジエステル体へと誘導する。オリゴヌクレオチドの合成では、ホスホロアミダイトモノマーの活性が高く極めて効率的に目的物を得ることができる12。一方で、この手法をグリコシルホスフェートの合成に応用する場合は、糖誘導体ならではの問題が生じる。中間体となるグリコシルホスファイトやホスホトリエステル体は、特に酸性条件下不安定で、アノマー位から容易にホスホン酸あるいはリン酸部位が脱離し得る(Figure 3)。実際、グリコシルホスファイトやホスホトリエステル体は、酸性条件下反応性が高いグリコシルドナーとして利用されている13,14。そのため、ホスホロアミダイト法をグリコシルホスフェートの合成に用いる場合には、これらの反応中間体の不安定性をいかに回避するかが課題となる。
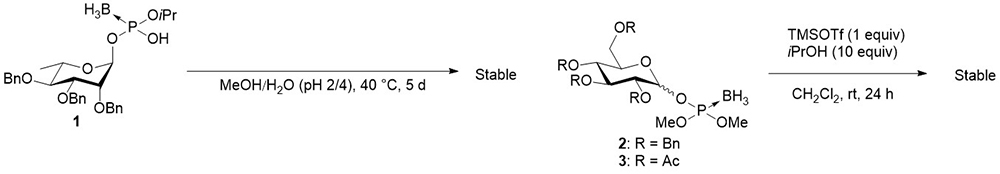
以上のような背景を踏まえ筆者らは、グリコシルボラノホスフェートを鍵中間体とする合成法に注目した。グリコシルボラノホスフェートはグリコシルホスフェートのリン酸部の非架橋酸素原子の一つをボラノ基に置換した化合物であり、リン原子の酸化数が+3のホスホン酸の誘導体である。Prosperiらは、このグリコシルボラノホスフェートのジエステル体1が、酸性の水-メタノール混合溶媒中で40 °Cに加熱したとしても、5日間全く分解しなかったことを報告している15。さらにMatsumuraらは、ボラノホスホトリエステル体2, 3が、対応するホスホトリエステルやホスファイトトリエステル体が即座にグリコシル化反応を起こすような、1当量のTMSOTfおよび10当量の2-プロパノール存在下の条件であっても、24時間で全く分解しないことを報告している16。このように、グリコシルボラノホスフェートは顕著に化学的安定性が高い誘導体であることが示唆されていた。特筆すべきは、ボラノホスホジエステル体は、トリチルカチオンと反応させることでH-ホスホネートジエステル体へと変換が可能である17。そこで、安定なボラノホスホトリエステル体を形成しながら鎖長を伸長し、合成の最終段階でH-ホスホネートジエステル体などの中間体を経てホスホトリエステル体へと変換できれば、効率的な合成法になりうると考えた。以降は、この戦略を用いた筆者らによる成果について紹介する。
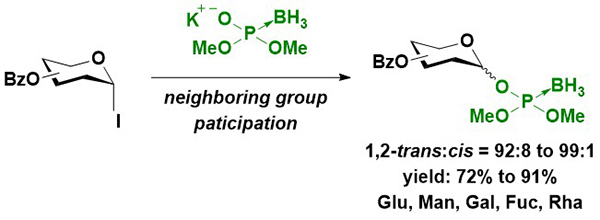
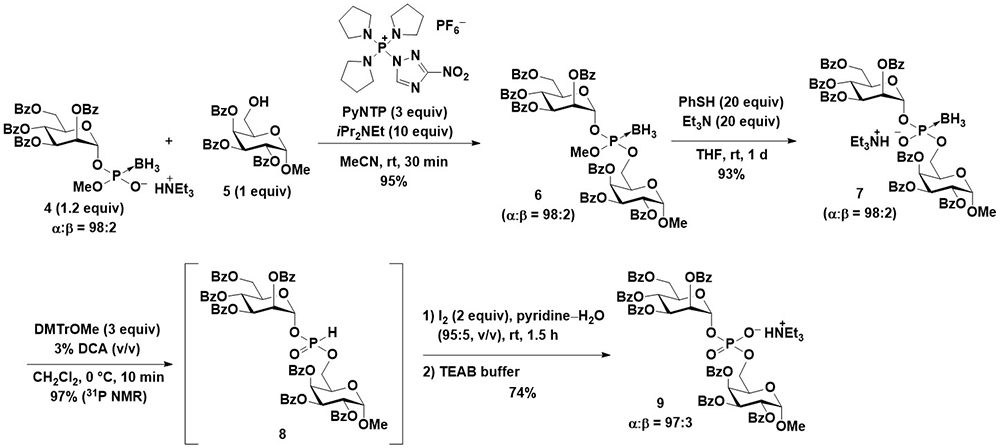
まず筆者は、グリコシルボラノホスホトリエステル体を、アノマー位の立体化学を制御して合成し、これを利用して糖鎖間のボラノホスホトリエステル結合の形成とホスホジエステル体への誘導によって、糖鎖間にリン酸結合を有する化合物を合成する戦略を試みた。Imamotoらの臭化糖を用いたアノマー位の立体化学を制御した合成例18を参考に、アシル系の保護基が導入されたヨウ化糖を用いてグリコシルボラノホスホトリエステルを1,2-トランス選択的に合成することに成功した(Figure 5)。この反応は2位のアシル基の隣接基関与効果によって立体選択性が発現していると考えられるが、中間体となる環状のジオキソカルベニウム中間体に対するヒドリド還元によってアセタールが生じる副反応が進行しやすい。水酸基の保護基としてアセチル基などの脂肪族カルボン酸エステル型のものを用いると前述の副反応が顕著に進行するが、芳香族カルボン酸エステル型の保護基を用いることで、この副反応の進行を抑制し、効率的にグリコシルボラノホスホトリエステルが得られた。続いて得られたボラノホスホトリエステル体の保護基を除去してジエステル体4とし(Figure 6)、これと糖水酸基との縮合反応を、縮合剤として筆者が所属する研究室で開発されたホスホニウム型縮合剤であるPyNTP19を用いて反応を行うことで、糖鎖間にボラノホスホトリエステル結合を有する2糖誘導体6を高収率で得ることに成功した。これをボラノホスホジエステル体7へと誘導後、酸性条件下DMTrカチオンと反応させてH-ホスホネートジエステル体8とし、ピリジン中ヨウ素と水を用いた酸化反応によってホスホジエステル体9へと変換することに成功した(Figure 6)。最終的なアノマー位の立体化学純度は、元のグリコシルボラノホスフェートのものと同等であり、立体特異的に合成が可能であることが示された20。
以上のように、ボラノホスフェートを合成中間体とすることでホスホジエステル体が合成可能であったが、最終的なボラノホスホジエステル体からホスホジエステル体への変換反応には課題があった。まずこの反応ではH-ホスホネートへの変換反応を酸性条件下で行い、その後のホスホジエステル体への誘導は塩基性条件下で行うため、一度抽出操作を行う必要があり、煩雑であった。そこで操作が簡便で、より広範なリン原子修飾体が合成可能な手法の開発に取り組んだ。リン-ホウ素結合は、リン原子周りの立体障害が大きいほど、さらには電子求引性基が存在しているほど、リン原子上の非共有電子対のホウ素に対する配位能が低下し、不安定化することが知られている21。そこで、ボラノホスホジエステル体に対して嵩高く、電子求引性が高い置換基を導入することで、リン-ホウ素結合を不安定化して脱ボラノ化によりホスファイト型の中間体を形成させ、これを起点にリン原子修飾グリコシルホスフェート誘導体の合成が可能になると考えた。
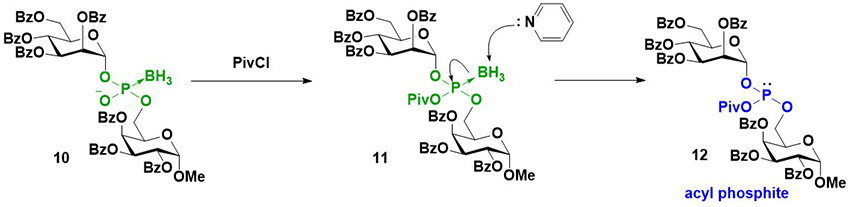
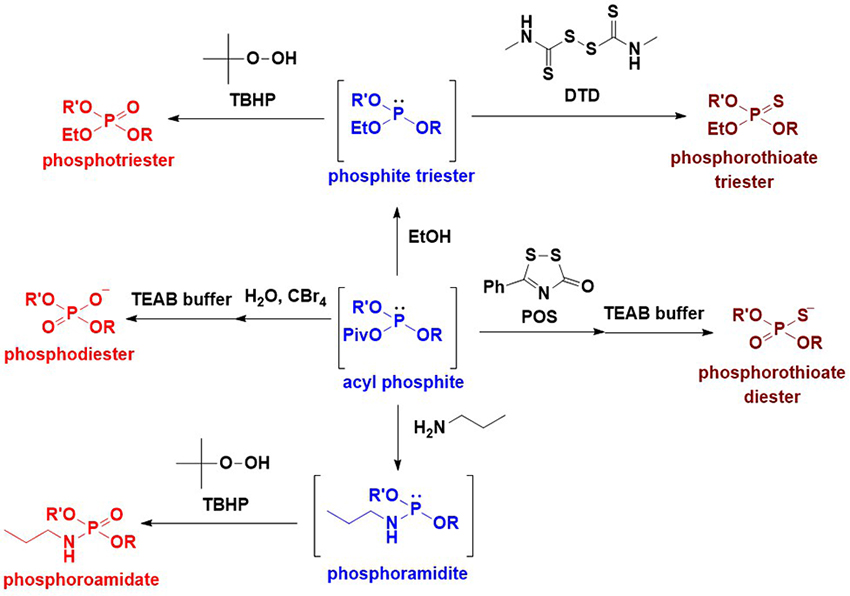
種々検討を行った結果、ピリジン中でピバロイルクロライドと反応させることで、ボラノホスホジエステル体がアシルホスファイトと呼ばれる誘導体へと効率的に変換されることを見出した。想定される反応機構をFigure 7に示す。ボラノホスホジエステル体10とピバロイルクロライドが反応して混合酸無水物11が得られる。嵩高く、電子求引性の高いピバロイル基の存在によってリン-ホウ素結合が弱まり、ピリジンによって脱ボラノ化することでアシルホスファイト12が生成したと考えられる。このアシルホスファイトは、例えば硫化剤である3-フェニル-1,2,4-ジチアゾリン-5-オン(POS)を共存させておくと、アシルホスファイトの非共有電子対がPOSと反応して硫化され、最終的にホスホロチオエートを定量的に与える。四臭化炭素存在下、アシルホスファイトを加水分解すると、H-ホスホネートを経由してホスホジエステル体へと変換される。さらには、アシルホスファイトをアルコール、アミンを反応させると、それぞれ対応するホスファイト、ホスホロアミダイトが生成する。これを酸化、または硫化することで、ホスホトリエステル、ホスホロアミデート、ホスホロチオエートトリエステル体などのリン原子修飾体を合成可能である(Figure 8)22。本手法は天然型であるホスホジエステル体を得ることができるだけでなく、ホスホロチオエートトリエステルのような、リン原子上に2重に修飾された誘導体も効率的に合成することが可能であり、グリコシルホスフェートだけではなく、オリゴヌクレオチドの種々のリン原子修飾体の合成にも応用されている23。
以上述べてきた通り、グリコシルボラノホスフェートを合成前駆体として糖鎖間の結合を形成し、これをホスホジエステル体を含め様々なリン酸誘導体へと変換可能な手法を開発してきた。そこで固相合成により、オリゴマーの合成を試みることとした。合成標的としてリーシュマニア原虫のLPGにみられる2糖1-ホスフェート繰り返し構造 [→6)-β-ᴅ-Gal-(1 → 4)-α-ᴅ-Man-(1-P)]6を選択し、適切な保護基を有する2糖1-ホスホロアミダイトビルディングブロックを用いて鎖長伸長を行うこととした。前述のように、ホスホロアミダイト法を用いる場合、中間体が不安定になる問題が生じるが、化学的に安定なボラノホスフェートの状態で鎖長を伸長し、合成の最終段階でホスホジエステル体へと変換すれば、効率的な合成が可能になると考えた。
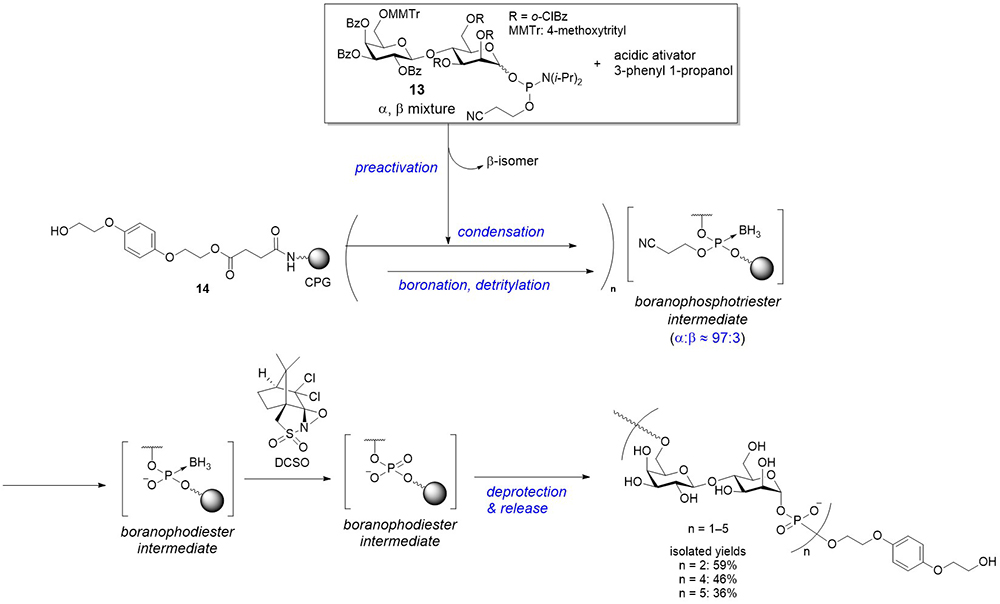
具体的な合成戦略をFigure 9に示す。。α:β比90:10程度の2糖1-ホスホロアミダイトをフェニルイミダゾールトリフラート(PhIMT)などの酸性活性化剤と、3-フェニル1-プロパノール存在下でプレアクティベーションを行い、その後controlled pore glass(CPG) 上の水酸基と縮合することでグリコシルホスファイトトリエステル体を形成する。このようなプレアクティベーションを行うのは、β体のビルディングブロックの方が反応性が高く、あらかじめアルコールと反応させて存在比を低減した状態で固相担体上の水酸基と反応させることで立体選択性が向上するためである24。次に生成したホスファイト中間体をBH3•SMe2によってボラノ化して化学的に安定なボラノホスホトリエステル体とし、続いてガラクトース6位に導入したMMTr基を酸性条件下除去する。その後再度プレアクティベーションを伴う縮合反応を行う、というサイクルを繰り返すことで鎖長を伸長する。目的の長さまで達したら、糖鎖間のボラノホスフェート構造をホスホジエステルへと変換する必要がある。ここで我々は、ボラノホスホトリエステル構造の保護基を除去して得られるボラノホスホジエステルは、オキサジリジンの誘導体であるDCSOと反応させることで、ある程度時間は要するもののホスホジエステルへ定量的に変換が可能であることを見出した。このような戦略を用いることで、最大5回繰り返し構造を有するグリコシルホスフェート誘導体を合成することに成功した(Figure 9)25。前述の様に、これまで合成がなされてきたグリコシルホスフェート誘導体は、鎖長の長さを制御したものではホスフェート構造を4か所有する誘導体が最長であり、最も長いオリゴマーを効率的に合成することに成功した。
前章ではグリコシルボラノホスフェートを合成前駆体としてグリコシルホスフェートを合成する手法について述べてきた。一方で、グリコシルボラノホスフェートをはじめ、リン原子修飾グリコシルホスフェートも創薬ターゲットや糖鎖の生体認識に関して知見を与えるプローブになりうる。例えばNikolaevの研究グループは、リーシュマニア原虫がLPGに有する2糖1-リン酸構造 [β-ᴅ-Gal-(1 → 4)-α-ᴅ-Man-(1-P)] のリン原子修飾体として、ボラノホスフェート、ホスホロチオエート、メチルホスホネート誘導体を合成した。この2糖1-リン酸構造はelongating Mannosyl Phosphate Transferase(eMPT)というリーシュマニアが有する固有の酵素によって認識され、非還元末端側のガラクトース6位がマンノシルホスホリル化されることが知られている。彼らは上記のリン原子修飾体が天然型と比べてどれだけマンノシルホスフェートを受容するかを調査し、メチルホスホネートでは全く受容能がなくなり、ボラノホスフェートでは50%、ホスホロチオエートでは75%程度の受容能を有することを報告している。電荷のないメチルホスホネートで全く受容能がなくなり、電荷のある誘導体では受容能があることから、リン酸あるいはホスホン酸構造にアニオンを有することが認識に極めて重要であることが示唆された26。このように、リン原子修飾体をプローブにして、酵素の認識に関する有用な知見を得ることが可能である。一方で、リン原子上に化学修飾を加えるとリン原子が不斉原子となり、立体異性体が生じる。こうした立体異性体は物理化学的、生物学的性質が大きく異なると考えられる。
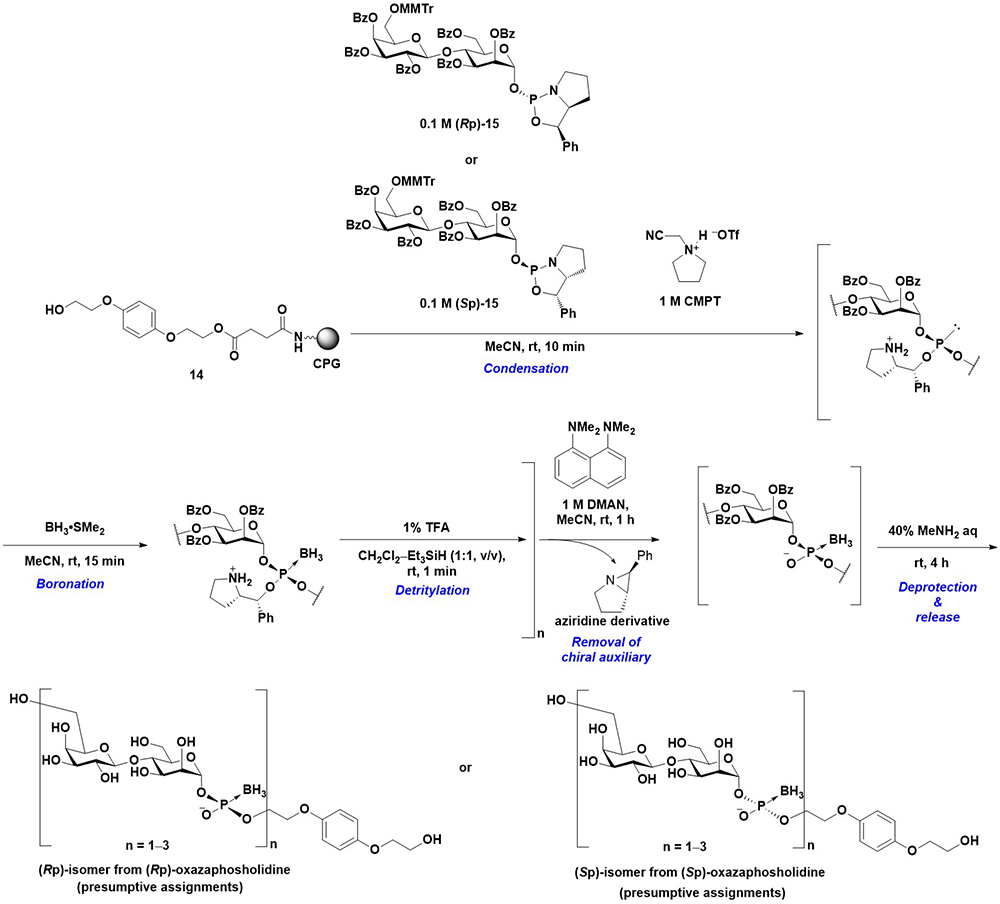
そこで、リン原子修飾グリコシルホスフェート誘導体を、リン原子の立体化学を制御して合成する研究に取り組んだ。リン原子の立体化学の制御には、オキサザホスホリジン法を用いることとした27。オキサザホスホリジンは不斉補助基を有する環状のホスホロアミダイト誘導体であり、あらかじめリン原子の立体化学が制御されたオキサザホスホリジンモノマーを、求核性のない酸性活性化剤存在下水酸基と反応させることで立体特異的に反応が進行し、立体化学が制御されたホスファイトトリエステル体が得られる28。さらに適切なリン原子修飾反応を行うことにより、様々なリン原子修飾体を立体選択的に合成可能である。まずは適切な保護基が導入された2糖1-オキサザホスホリジンモノマー15を合成した。詳細な理由は不明であるが、オキサザホスホリジンモノマーはα体のみを比較的容易に単離可能である。続いてCPG上の水酸基との縮合反応を、酸性活性化剤として求核性の低い1-(シアノメチル)ピロリジニウムトリフラート(CMPT)28存在下行い、ホスファイトトリエステル中間体を得る。この中間体をボラノ化したのち、酸性条件下でガラクトース6位に導入されたMMTr基を除去する。以上の縮合、ボラノ化、脱MMTr化のサイクルを繰り返すことで鎖長を伸長し、合成の最終段階で1,8-(ジメチルアミノ)ナフタレン(DMAN)を用いることで不斉補助基を除去し、ボラノホスホジエステル体とする。固相担体からの切り出しと水酸基の脱保護をメチルアミン処理によって行うことで、グリコシルボラノホスホジエステル誘導体を立体選択的に、最長3回繰り返し構造まで合成することに成功した(Figure 10)。目的物のNMRによる立体化学の帰属には至らなかったが、Rp、Sp体のグリコシルボラノホスフェートは、それぞれRp、Sp体のオキサザホスホリジンモノマーから得られたと考えられる。この合成の鍵となるのは以下の2点である。
(1) 不斉補助基の除去の際、求核性が極めて低いDMANを用いることが極めて重要である。この工程は、不斉補助基のピロリジン環を塩基によって活性化し、アジリジン誘導体を形成しながら不斉補助基が除去される。より求核性の高い塩基を用いてしまうと、アジリジン誘導体の形成よりも脱ボラノ化が進行してしまうため、目的物の収率が大きく低下する。
(2) ボラノ化後にエタノールによって固相担体を洗浄することが有効である。ボラノ化剤、およびその残渣が残存した状態で縮合反応を行うと、著しく反応効率が低下するが、固相担体をエタノールで洗浄することでこうしたボラノ化剤やその残渣が除去され、縮合効率が向上する。
同様に、ボラノ化のステップを硫化に置き換えることで、グリコシルホスホロチオエートを立体選択的に合成することも可能である29。このような立体化学が制御されたリン原子修飾グリコシルホスフェート誘導体は、生体内での糖鎖の機能解明に向けた有用なプローブになることが期待される。
これまで述べてきたように、グリコシルボラノホスフェートを合成中間体とすることで、グリコシルホスフェート構造を効率的に合成することに成功した。さらに、グリコシルボラノホスフェートをホスフェートだけではなく、種々のリン原子修飾体へと変換を可能な反応の開発にも成功した。
グリコシルボラノホスフェートは合成前駆体としてだけでなく、リン原子修飾アナログとしてその生理活性は極めて興味深く、創薬ターゲットになり得る分子である。そこで、リン原子のキラリティーを制御した合成にも取り組んだ。これまでに開発された方法を医薬品やワクチン開発に繋げるべく、今後も研究を続ける所存である。



