|
|
Stereoselective Synthesis of 1,2-cis-Glycosides |
 |
|||||||||||||||||||||||||||||||||||||||||||
 |
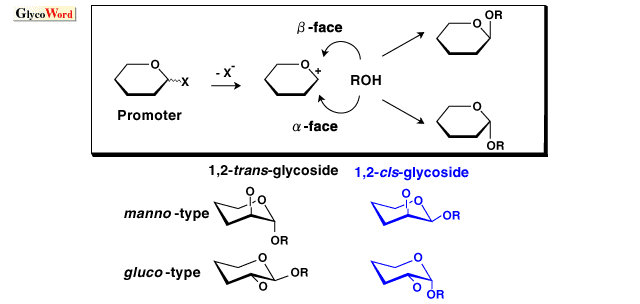
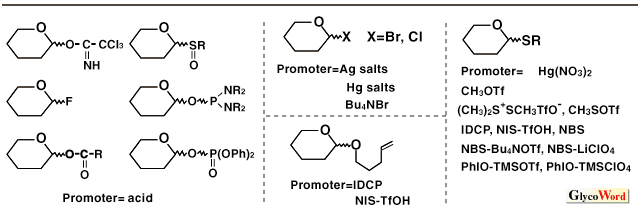
Synthesis of oligosaccharides and glycoconjugates has become increasingly important for elucidation of their biological functions. Stereoselective glycosylation has been a major issue for efficient synthesis of oligosaccharides and glycoconjugates (1,2). Glycosylation reaction generally proceeds as follows: activation of a glycosyl donor by an appropriate promoter followed by elimination of a leaving group at the 1-position affords an oxocarbenium ion intermediate; a hydroxy group of a glycosyl acceptor then attacks the oxocarbenium ion intermediate to form a glycosidic bond. Addition of the hydroxy group from the upper face of the pyranose (or furanose) ring of the oxocarbenium ion forms beta-glycoside, whereas the addition from the lower face forms alpha-glycoside. Glycosylation with 2-O-acylated donors usually affords 1,2-trans-glycoside stereoselectively by virtue of neighboring group participation of the 2-O-acyl group. For preparation of 1,2-trans-glycoside of 2-amino sugars such as glucosamine and galactosamine, neighboring group participation can be employed by using 2,2,2-trichloroethoxycarbonyl and phthaloyl groups for protection of amino function. (Glycosylation with N-acetyl sugar also affords 1,2-trans-glycoside via oxazoline intermediate.) Stereoselective formation of 1,2-cis-glycosides and 2-deoxyglycosides (and alpha-sialosides) is generally a difficult issue where no assisting effect such as participation of a neighboring group is available. Ether-type protective groups (benzyl, allyl, etc.) are used for protection of 2-hydroxy function in order to form 1,2-cis-glycosidic bonds. 2-Azido-2-deoxy sugars are used as precursors for preparation of 1,2-cis-glycosides of 2-amino sugars. 1,2-cis-Glycosides are categorized to alpha-gluco-type and beta-manno-type where the 2-hydroxy group is equatorial and axial, respectively. Since the alpha-anomer is thermodynamically more stable than the beta-anomer by anomeric effect, 1,2-cis-alpha-glucoside is preferentially obtained in general. Preparation of 1,2-cis-beta-mannoside is particularly difficult since the formation of 1,2-cis-beta-mannoside is unfavorable thermodynamically as well as kinetically owing to steric hindrance of axial 2-hydroxy group. Previously, beta-mannosides were only able to synthesize via SN2-like reaction of alpha-glycosyl halide by using insoluble silver salts. Various studies have been conducted for beta-mannosylation. Recently, Hindsgaul, Stork, Ito and Fairbanks reported stereoselective beta-mannosylation by intramolecular aglycon delivery (3,4). In this method, the glycosyl acceptor is delivered to the donor via a linker attached on the donor moiety. Ito's method is one of the most practical beta-mannosylation so far as reported both yield and selectivity. beta-Mannosylation was also effected by intramolecular glycosylation using a "molecular clamp," which connects a donor to an acceptor to kinetically accelerate the glycosylation and allows a stereo- and regioselective reaction by controlling the spatial arrangement of a donor and an acceptor (5). [beta-D-Manp-(1 Many methods for alpha-selective glycosylation of the gluco-type have been published but they are not universal. Lemieux's in situ anomerization using glycosyl halides as donors and Bu4NBr as a promoter was already reported in the mid 1970s. Konigs-Knorr method using silver salts or mercury salts as promoters has also been used. Among various methods reported for selective alpha-glycosylation in recent years, the combinations of diethyl ether as a solvent and perchlorates as a source of counter anion against oxocarbenium ion have frequently been used. Recently, alpha-orienting solvent effect of dioxane and t-butylmethyl ether was also reported. Glycosyl fluorides proved to be versatile glycosyl donors for alpha-glycosylation. Lewis acids such as SnCl2-AgClO4 (Mukaiyama method), TMSOTf (Noyori method: trimethylsilylated acceptor is used), and Cp2ZrCl2-AgClO4 (Suzuki method) have been used as promoters for alpha-selective glycosylation. New methods for alpha-glycosylation using 1-O-acyl sugar, glycosyl phosphate, 1-OH sugar, and others have been reported. Precise assessment of the utility of these new methods are under way. Thioglycosides have been recognized as versatile glycosyl donors particularly for synthesis of complex carbohydrates since they are stable under a variety of reaction conditions but can be activated with appropriate thiophilic reagents. Various methods for activation of thioglycosides have been reported: promoters such as iodonium dicollidine perchlorate (IDCP) (1), NBS-LiClO4, NBS-LiNO3, NBS (NIS) -AgClO4, PhIO-TMSClO4, PhIO-SnCl4-AgClO4, N-phenylselenophthalimide-Mg(ClO4)2 are useful for alpha-selective glycosylation (7,8,9). Pentenyl glycosides were also used as glycosyl donors for alpha-glycosylation using IDCP as a promoter. In order to obtain alpha-glycoside stereoselectively, reactivities of glycosyl donor and promoter should be taken into consideration. Highly reactive glycosyl donors are not suitable for alpha-selective glycosylation. Reactivity of the glycosyl donor can be controlled by protective groups. Electron-withdrawing groups such as acyl groups decrease reactivity, whereas electron-donating groups such as alkyl and silyl groups increase reactivity. For example, glycosylation with 3,4,6-tri-O-acetyl-2-O-benzyl glycosyl donor tends to give alpha-glycoside more selectively than that with a perbenzylated donor. Glycosyl trichloroacetimidates, which proved to be one of the most practical glycosyl donors are not suitable for alpha-selective glycosylation in general because of their high reactivity. Glycosylation with 2-azido-2-deoxyglucosyl trichloroacetimidate, however, affords alpha-glycoside with high selectivity by using TBDMSOTf as an activator (10). Steric hindrance also greatly influences stereoselectivity. Glycosylation with 2-O-benzylated galactosyl and fucosyl donors, which have an axial 4-hydroxy group, tend to give alpha-glycosides with high selectivity. The bulky protective groups (TBS, Trt, TBDPS) introduced at the 6-position of glucosyl donors increase alpha-selectivity (9,11). 6-O-Dinitrobenzoyl, carbamoyl and Troc groups also increase alpha-selectivity, probably owing to participation of the carbonyl function of the 6-O-protective groups to oxocarbenium ion from the beta-face (8). More secure alpha-selective glycosylation is sometimes required, since the above methods are not always effective enough. Intramolecular glycosylation methods such as intramolecular aglycon delivery and the molecular clamp method also proved to be useful for secure formation of alpha-glycosic bonds (12, 13, 14). Although these methods require additional reaction steps for linking glycosyl donor with glycosyl acceptor, these methods are expected to be useful particularly for complex oligosaccharide synthesis via segment condensation. |
|||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
| Koichi Fukase (Department of Chemistry, Graduate School of Science, Osaka University) | ||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||
| Dec. 15, 1999 | ||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||