|
|
Reaction Mechanism of Glycosylase |
 |
||||||||||
 |
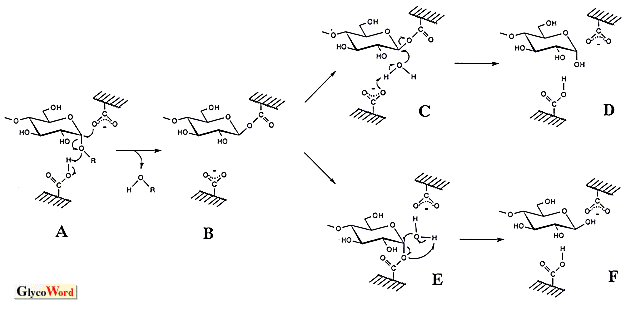
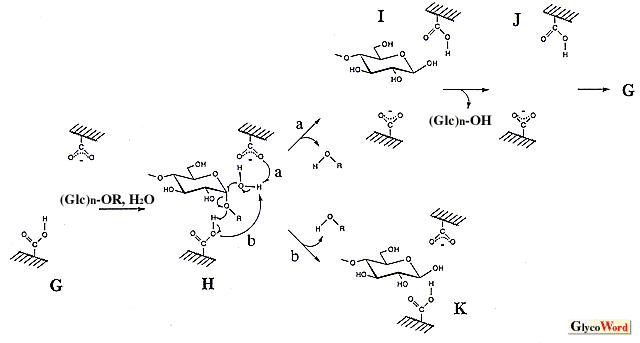
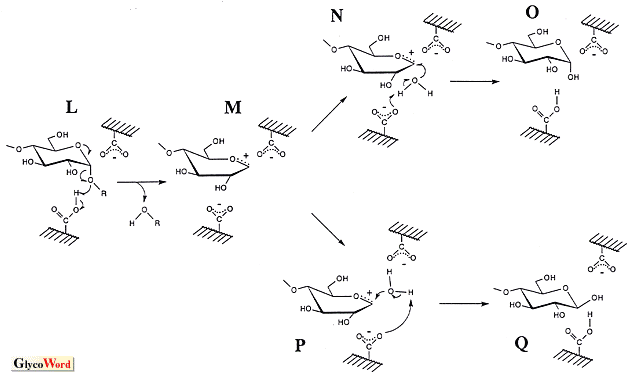
Glycosylase is an enzyme group that hydrolyzes the glycosyl linkages in oligosaccharide, polysaccharide and sugar conjugate. Taking the topic of this series into consideration, this manuscript focuses on the amylase which cleaves the bond between anomeric carbon and glucosyl oxygen atoms. These enzymes catalyze hydrolysis, transglycosylation and condensation in which the last two reactions produce the glycosyl oligomer of high DP. They have been utilized to synthesize oligosaccharides with excellent physiological functions. Transglycosylation is a reaction that transfers the glycosyl residue into the alkyl (or aryl) hydroxyl group in the acceptor, and its mechanism is considered to be the same as that of hydrolysis, implying that the hydrlysis is a kind of transfer reaction into water. Therefore, the substrate (donor) for the hydrolytic reaction is essential to transglycosylation, and various kinds of products are obtained by the appropriate selection of the acceptor. If a high yield is desirable, the acceptor concentration should be high, since transglycosylation competes with hydrolysis. Condensation is the reverse of hydrolysis, and the product of hydrolysis is the starting material (substrate) for this reaction. The anomeric configuration of the glycosyl linkage formed by transglycosylation is identical to that by hydrolysis, and in condensation it becomes the configuration of the substrate of hydrolysis. The reasonable hydrolytic reaction mechanism of amylase remains to be established. There seem to be two significant models, an SN1-type mechanism via oxocarbenium ion intermediate and an SN2-type mechanism with nucleophilic displacement. Experimental data supporting both of them have been obtained. According to the anomeric configurations of substrate and product, amylases are classified into two groups, i.e., retaining enzymes such as alpha-amylase and alpha-glucosidase and inverting enzymes such as beta-amylase and glucoamylase. To explain the retaining and inverting reaction, two mechanisms give the schemes shown in the figure. (The catalytic ionizable groups of amylases seem to be carboxylate and carboxyl. The imidazole was formerly considered to be a catalytic group of alpha-glucosidase, but is actually a protonated carboxyl, carboxy.) The retaining and inverting mechanisms of the anomeric configuration in the SN2-type reaction may be considered individually. In the case of the retaining enzyme, the -COO- attacks the anomeric carbon atom with cooperative cleavage of the glucosyl linkage by -COOH (A in the figure). After the formation of the glucosyl enzyme via covalent bond (B), the second attack of the water molecule (C) retains the anomeric configuration (D). If this scheme is applied to the inverting enzyme, assuming that the SN1 and SN2 mechanisms can proceed in overlapping routes, another -COO- reacts on the anomeric carbon atom before the attack of water (E), indicating triple displacement. Such displacements seem unlikely to occur in a hydrolytic reaction. Therefore, another mechanism has been proposed to complete the reaction by a one-time nucleophilic substitution (H). In the case of the SN1-type reaction, after the split of glucosyl linkage by -COOH (L) the oxocarbenium ion intermediate formed was stabilized by a negatively charged carboxyl group (M, formation of ionic bond). The mechanism of the anomeric retention and inversion was interpreted simply by the direction of OH- addition (N and P, respectively). In the scheme for the inverting-type enzyme the structure of I is shown at the end of the reaction, which implies that the activation of the water molecule is done by -COO-, reversing the protonation of two carboxyl groups before and after hydrolysis (see G and J). Before the reaction on new substrate, the proton movement (J to G) is essential (if following arrow b in H, K is formed and the structure of G is restored). In the SN2-type triple displacement mechanism and the SN1-type mechanism, if the proton of water is pulled out by -COO- (E and P, upper one in P), the same reversion of protonation occurs and the proton movement is needed. There is a proposal that the proton replacement is done between carboxyl groups and water in solution according to the pKe values. A comparison of the SN1-type anomer-retaining mechanism with the SN2-type shows a clear difference in the linkage between the carboxyl group and the intermediate (oxocarbenium cation in SN1, glucosyl group in SN2), that is, an ionic bond for SN1 and a covalent bond for SN2, suggesting that the distance of the linkage is important. When a perfect ionic or covalent linkage is formed, the bond distance should be one of both linkages. There is an opinion that doubts such a perfect formation in which the linkage has ionic and covalent bond-like characteristics if the distance is between them (a natural concept of chemical linkage), suggesting the possibility of an integrated reaction mechanism of anomer-retaining amylase. The bond distance in each elementary reaction step may change, and the experimental data supporting SN1-type and SN2-type reaction mechanisms can be explained by this. On one hand, SN1 and SN2 mechanisms of the reverting reaction are distinguished. As shown in H, the nucleophilic attack of an activated water molecule on the anomeric carbon atom occurs in concert with the reaction of -COOH on the oxygen atom in the glycosyl bond to complete the hydrolysis at once, a fact which does not permit an intermediate such as M. A recent review (J. Appl. Glycosci. 46, 187-197, 1999) published by Kaneko et al. is recommended for study of the reaction mechanisms simulated by modeling and docking. It is difficult to detect directly the intermediate state in enzyme reactions of very short duration. Kaneko et al. applied an "effective computer simulation" method to investigate this and interpreted interesting findings. |
||||||||||
|
|||||||||||
| Atsuo Kimura (Graduate School of Agriculture, Hokkaido University) | |||||||||||
|
|||||||||||
| Sep. 15, 1999 | |||||||||||
|
|
|||||||||||
|
|||||||||||