|
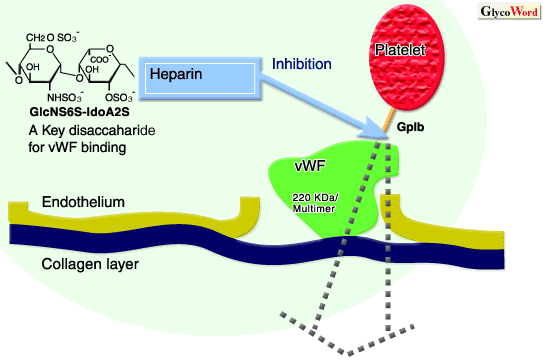
The protein von Willebrand factor (vWF) is a large cysteine-rich glycoprotein with a subunit mass of approximately 220 kDa. Through disulfide bonds, vWF forms multimeric assemblies composed of 2 to 50 subunits. This protein is synthesized in vascular endothelial cells or megakaryocytes, and secreted into the subendothelial matrix or stored in the a-granules of platelets differentiated from megakaryocytes. vWF plays two essential roles in hemostasis. First, it circulates in the plasma as a complex with Factor VIII to stabilize and protect it from inactivation by activated Protein C. If the Factor VIII is not in the complex with vWF, Factor VIII disappears from plasma in only minutes. Second, vWF assists primary platelet hemostasis via the formation of the platelet plug. That is, when vascular injury occurs and the subendothelial extracellular matrix of the vessel wall is exposed to the flowing blood, vWF is a critical protein to which platelets initially adhere. Normally, vWF in the circulating blood assumes a low affinity conformation that does not bind to platelets. But when the protein attaches to (or is exposed within) the extracellular matrix, it changes conformation. This change in conformation primarily affects the high affinity A1 domain in vWF, which binds to its platelet receptor, glycoprotein Ib (GpIb). Tight binding between vWF and GpIb then occurs. This binding is also induced by high shear flow, as can be seen in narrowed arteries. In the case of vWF-platelet binding induced by high shear, it is not precisely known whether the vWF protein, or the GpIb receptor, or both undergo changes in affinity. Once platelets adhere to vWF, platelet-platelet aggregation is further induced and enhanced through the integrin GpIIb/IIIa receptors on platelets, through binding to fibrinogen (and secondarily to vWF). Therefore, the binding between vWF and GpIb, and the resulting platelet adhesion to injured vessels is very important for primary hemostasis - platelet plug formation. But when these physiological processes go beyond normal hemostatic mechanisms, the accumulation of platelets at sites of vascular injury may contribute significantly to the early and late failures of many cardiovascular interventions. Thus, much research has been committed to discovering ways of controlling these initial events of platelet-dependent hemostasis.
In 1991, it was first demonstrated that the intravenous administration of heparin to patients during open heart surgery induced the impairment of vWF-dependent platelet function, without changes in plasma vWF levels (1). This inhibitory effect of heparin on vWF-dependent platelet agglutination was not dependent on the heparin's affinity for antithrombin III, but was dependent upon the molecular weight of heparin. From later in vitro experiments, it was found that heparin bound to a specific amino acid sequence within the A1 domain of vWF (residues 569 - 583), in which basic amino acids are regularly arranged. Heparin binding induced conformational changes in a peptide of this binding site (2). Heparin bound to both activated and inactivated vWF similarly, but did not interfere with vWF binding to collagen. Since the platelet GpIb-binding domain (residues 524 - 542) is also located in the A1-loop, it was suggested that heparin interferes with vWF binding to platelet GpIb both by steric hindrance and by inducing a conformational change of the domain.
Regarding the structural specificity of heparin that is responsible for binding to vWF, a key disaccharide unit (GlcNS6S-IdoA2S, see figure) was identified. This structural unit, which is normally destroyed by heparinase I digestion, was successfully deduced by competitive binding assays using heparin fractions prepared by specific methods of depolymerization of heparin that produce fragments of predictable structure (3). Further, through the study using synthetic and structurally defined oligosaccharides, it was demonstrated that the assembly of more than 3 units of the disaccharide was crucial for the binding potency.
| |
|
| References | (1) | Sobel M, McNeill PM, Carlson PL, Kermode JC, Adelman B, Conroy R, Marques D, J. Clinic. Invest. 85, 1787-1793, 1991
|
|
(2) |
Sobel M, Soler DF, Kermode JC, Harris RB, J. Biol. Chem. 267, 8857-8862, 1992
|
|
(3) |
Poletti PL, Bird KE, Marques D, Harris RB, Suda Y, Sobel M, Arterioscler Thromb. Vasc. Biol. 17, 925-931, 1997
|
| |