|
Lipid-linked oligosaccharide (Glc3Man9GlcNAc2-P-P-Dol) serves as a precursor to the asparagine-linked oligosaccharide of glycoprotein.
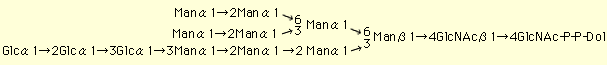
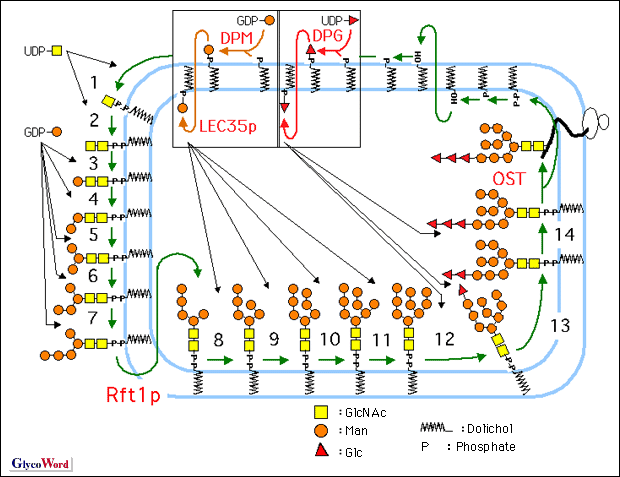
The synthesis of lipid-linked oligosaccharide is initiated on the cytoplasmic face of the endoplasmic reticulum (ER) membrane, where two N-acetylglucosamine (GlcNAc) residues and five mannose residues (Man) are assembled using UDP-GlcNAc and GDP-Man, respectively, as sugar donors. After translocation of the Man5GlcNAc2-P-P-Dol to the luminal face of the ER membrane, four mannose residues and three glucose residues transfer from Dol-P-Man and Dol-P-Glc, respectively, as sugar donors. The process is highly conserved among eucaryotic cells (1).
The biosynthesis is dependent on the supply of Dol-P (de novo synthesis and recycling) (2), sugar donors (nucleotide sugars and dolichol-linked sugars) and acceptors, the activities of 14 separate glycosyltransferases, and the presence of proteins, called flippases, Rft1 for Man5GlcNAc2-P-P-Dol (3), LEC35 protein for Dol-P-Man, and the LEC35-like protein for Dol-P-Glc (4). Moreover, these activities are regulated at the transcriptional level, enzymatic level, and so on.
Congenital disorders of glycosylation (CDGs) are human inherited metabolic diseases resulting from the aberrant synthesis of the asparagine-linked oligosaccharide of glycoproteins. Diagnosis is based on the clinical findings of psychomotor and mental retardation, and liver problems, combined with the biochemical finding of an abnormally high isoelectric point of serum glycoproteins (such as transferrin), reflecting the deficiency of sialic acid. CDGs are divided into two subtypes, type I (CDG-I) and type II (CDG-II), based on the differences in the biochemical defect. CDG-I cases are biochemically diagnosed by the deficiency of asparagine-linked oligosaccharide of glycoprotein (underglycosylation), and CDG-II cases are diagnosed by the abnormal processing of the asparagine-linked oligosaccharide of glycoprotein (5).
Underglycosylation of CDG-I cases can result from defects in both the supply of the lipid-linked oligosaccharide and the oligosaccharyltransferase activity. In fact, seven different subtypes, designated CDG-Ia to g, have been identified in CDG-I to date. There are two subtypes that have a defect in the synthesis of sugar phosphate. If one study, CDG-Ia patients (about 350 cases) showed a defect in the PMM2 gene that encodes phosphomannomutase 2 (Man-6-P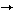 Man-1-P), and CDG-Ib patients (about 30 cases) showed a defect in the PMI gene that encodes phosphomannose isomerase (Fru-6-PMan-6-P). CDG-Ib patients are treatable with mannose administration. The other subtypes (1 to 12 cases, respectively) showed defects in the assembly of lipid-linked oligosaccharide: CDG-Ic patients had a defect in the hALG6 gene that encodes Man-1-P), and CDG-Ib patients (about 30 cases) showed a defect in the PMI gene that encodes phosphomannose isomerase (Fru-6-PMan-6-P). CDG-Ib patients are treatable with mannose administration. The other subtypes (1 to 12 cases, respectively) showed defects in the assembly of lipid-linked oligosaccharide: CDG-Ic patients had a defect in the hALG6 gene that encodes  1-3 glucosyltransferase (glucosyltransferase I, Fig.1: step 12 ), and showed accumulation of Man9GlcNAc2-P-P-Dol. CDG-Id patients had a defect in the hALG3 gene that encodes mannosyltransferase VI (Fig.1: step 8), and showed accumulation of Man5GlcNAc2-P-P-Dol. CDG-Ie patients had a defect in the DPMI gene that encodes Dol-P-Man synthase (Fig.1: DPM) subunit Dpm1p, and showed accumulation of Man5GlcNAc2-P-P-Dol. CDG-If patient had a defect in the MPDU-1/LEC35 gene that encodes Dol-P-Man flippase, LEC35 protein (Fig.1: LEC35p), and showed accumulations of Man5GlcNAc2-P-P-Dol, Man9GlcNAc2-P-P-Dol, etc. CDG-Ig patient had a defect in hALG12 gene that encodes mannosyltransferase VIII (Fig.1: step 10), and showed accumulation of Man7GlcNAc2-P-P-Dol. Until this investigation, CDG-Is that showed a defect in the first half of the assembly of lipid-linked oligosaccharide had not been detected. 1-3 glucosyltransferase (glucosyltransferase I, Fig.1: step 12 ), and showed accumulation of Man9GlcNAc2-P-P-Dol. CDG-Id patients had a defect in the hALG3 gene that encodes mannosyltransferase VI (Fig.1: step 8), and showed accumulation of Man5GlcNAc2-P-P-Dol. CDG-Ie patients had a defect in the DPMI gene that encodes Dol-P-Man synthase (Fig.1: DPM) subunit Dpm1p, and showed accumulation of Man5GlcNAc2-P-P-Dol. CDG-If patient had a defect in the MPDU-1/LEC35 gene that encodes Dol-P-Man flippase, LEC35 protein (Fig.1: LEC35p), and showed accumulations of Man5GlcNAc2-P-P-Dol, Man9GlcNAc2-P-P-Dol, etc. CDG-Ig patient had a defect in hALG12 gene that encodes mannosyltransferase VIII (Fig.1: step 10), and showed accumulation of Man7GlcNAc2-P-P-Dol. Until this investigation, CDG-Is that showed a defect in the first half of the assembly of lipid-linked oligosaccharide had not been detected.
Most of the genes used for assembly of the lipid-linked oligosaccharide have been isolated from Saccharomyces cerevisiae (1). However, a few genes remain to be isolated. Using the nucleotide sequence of the yeast genes, corresponding mammalian genes, including human genes, have been isolated by homology cloning. We have succeeded in isolating human genes encoding GDP-Man-dependent mannosyltransferases (mannosyltransferase I to V) (6-8).
As described above, the synthetic pathway and its regulation of lipid-linked oligosaccharide is very complicated; novel subtypes of CDG-I will be detected in parallel with their precise analysis.
|
|
|
| References |
(1) |
Burda P, Aebi M, Biochim. Biophys. Acta 1426, 239-257, 1999 |
|
(2) |
Schenk B, Fernandez F, Waechter CJ, Glycobiology 11, 61R-70R, 2001 |
|
(3) |
Helenius J, Ng DTW, Marolda CL, Walter P, Valvano MA, Aebi M, Nature 415, 447-451, 2002 |
|
(4) |
Anand M, Rush JS, Ray S, Doucey M-A, Weik J, Ware FE, Hofsteenge J, Waechter, C.J. and Lehrman, M.A. Mol. Biol. Cell 12: 487-501 (2001) |
|
(5) |
Jaeken J, Carchon H, Curr. Opin. Neurol. 14, 811-815, 2001 |
|
(6) |
Kataoka K, Takahashi T, Ayusawa D, Nishikawa Y, Somat. Cell Mol. Genet. 24, 235-243, 1998 |
|
(7) |
Takahashi T, Honda R, Nishikawa Y, Glycobiology 10, 321-327, 2000 |
|
(8) |
Takahashi T, Nishikawa Y, Glycoconjugate J. 18, 118, 2001 |
|
|
|
|
|