 |
プロテオグリカン (PG) は糖側鎖としてグリコサミノグリカン (GAG) を有し、細胞表面や細胞外マトリックスにおいて、シグナル伝達の調節や細胞外基質の構築を担っている(図 1)(1-5)。PGを構成するGAGにはコンドロイチン硫酸 (CS)、デルマタン硫酸 (DS)、ヘパラン硫酸 (HS)、ケラタン硫酸 (KS) が含まれ、その側鎖の違いでCS-PG、DS-PG、HS-PG、KS-PGに分類される。CS、DS、HS、KSは、それぞれ軟骨、皮膚・大動脈、肝臓・腎臓、角膜・軟骨に多く含まれる。PGのコアタンパク質部分をコードする遺伝子の変異によって、骨格異常、神経異常、心疾患、眼・耳疾患を発症する(表 1)(6)。CS/DS/HS/KSの生合成機構と遺伝性疾患については、本シリーズの他の項を参照されたい。
1. コンドロイチン硫酸プロテオグリカン欠損症
ACANによってコードされるアグリカンは、2,431アミノ酸から構成され、1分子あたり100本以上のCSとKS、N-グリカンから成る。ACANの変異によって、常染色体顕性(優性)のキンバリー型/アグリカン型の脊椎骨端異形成症が発症する (7-9)。本疾患の特徴は、低身長、ずんぐりとした体格、早期発症の関節変形性関節症、硬化症と終板の不規則性を伴う平らな椎体、扁平化した大腿骨の骨端、離断性骨軟骨炎などである。
VCANによってコードされるバーシカンは、3,396アミノ酸から構成され、1分子あたりおよそ20箇所の推定CS付加部位とO-グリカンおよびN-グリカンから成る。VCANの変異によって、常染色体顕性のワーグナー症候群が発症する (10-12)。本疾患の特徴は、硝子体網膜症、硝子体腔の空洞、硝子体変性、進行性脈絡網膜萎縮、血管周囲鞘形成、嚢下白内障、近視などである。
2. デルマタン硫酸プロテオグリカン欠損症
BGNによってコードされるビグリカンは、368アミノ酸から構成され、1分子あたり2箇所のDS付加部位をもつ。BGNの変異によって、X染色体潜性(劣性)の脊椎骨端骨幹端異形成症が発症する (13)。本疾患の臨床症状の特徴は、脊椎の異形成、長骨の骨端と骨幹端の異形成に起因する低身長や変形性関節症である。また、BGNの別の変異によって、メーステル・ロイス症候群(胸部大動脈瘤・解離)を発症する (14)。本疾患は、早発症の大動脈瘤および大動脈解離、関節の可動域亢進および拘縮、皮膚線条および胸郭の変形、口蓋垂開裂および頸椎の不安定症、頭蓋顔面異形症(長頭症、両目隔離症、斜視、高眼瞼、眼球突出、頬骨低形成、前頭隆起)を特徴とする。メーステル・ロイス症候群は、トランスフォーミング増殖因子-β (TGF-β) およびTGF-β受容体の変異に起因するロイス・ディーツ症候群やフィブリリン1 (FBN1) の変異に起因するマルファン症候群、3型コラーゲンの変異に基づく血管型エーラス・ダンロス症候群の症状と似ていることから、ビグリカンあるいは側鎖のDSがTGF-βのシグナル伝達の調節やコラーゲン線維束の形成に寄与していると考えられる。実際に、ビグリカンはin vitroでTGF-βと結合し、DSはコラーゲン線維と結合し束ねる役割を有する。
DCNによってコードされるデコリンは、359アミノ酸から構成され、1分子あたり1箇所のDS付加部位をもつ。DCNの変異によって、常染色体顕性の角膜実質ジストロフィーが発症する (15-20)。興味深いことに、報告されている患者の変異は全てデコリンタンパク質が短くなる変異で、一アミノ酸置換を引き起こす変異はこれまで報告されていない。おそらくこの変異型デコリンがドミナントネガティブ体として機能するのであろう。角膜実質ジストロフィーの臨床症状の特徴は、小さな薄片や斑点として観察される微細混濁を伴う角膜混濁、屈折異常、弱視、斜視、眼振、内斜視などである。
3. ヘパラン硫酸プロテオグリカン欠損症
ヒトにおいてグリピカンファミリーには6種類が知られており、GPC1〜GPC6によってコードされるグリピカン1〜6は、その進化系統樹やアミノ酸配列の相同性(およそ25%)から、GPC1/2/4/6とGPC3/5のサブファミリーに分類されている。グリピカンファミリータンパク質は、550〜800アミノ酸から構成され、1分子あたり推定2箇所のHS付加部位をC末端側にもち、グリコシルホスファチジルイノシトール (GPI) アンカー型として細胞表面に存在している。GPC3の変異によって、X染色体潜性の1型シンプソン・ゴラビ・ベーメル症候群が発症する (21-25)。本疾患の臨床症状の特徴は、出産前後の過成長、内臓および骨格の異常、粗い顔立ち、心臓欠陥、低血圧などである。GPC4の変異によって、X染色体劣性のケイパート症候群(鼻・指・聴覚症候群)が発症する (26, 27)。ケイパート症候群の臨床症状の特徴は、頭蓋顔面および指の形成異常、学習障害、感音性難聴などである。GPC6の変異によって、常染色体潜性の肩異形成症が発症する (28, 29)。本疾患の臨床症状の特徴は、四肢近位短縮、顔面異形成、重度の低身長、上腕骨低形成などである。
HSPG2によってコードされるパールカンは、4,392アミノ酸から構成され、1分子あたり2〜3箇所の主にHS付加部位をもつが、CSの場合もある。基底膜に多く存在しているが、筋肉や軟骨、骨髄の細胞外マトリックスにも豊富である。HSPG2の変異によって、常染色体潜性の1型シュワルツ・ヤンペル症候群(軟骨異栄養性筋強直症)あるいはシルバーマン・ハンドマーカー型またはローランド・デビュクオア型分節異常骨異形成症が発症する (30-38)。
1型シュワルツ・ヤンペル症候群(軟骨異栄養性筋強直症)の臨床症状の特徴は、筋弛緩の長期不全ミオトニアおよび低身長、脊柱後弯症、骨幹部の弯曲、骨端の不整などの骨格形成異常を示す (30-35)。ミオトニアとは、筋の持続収縮および弛緩障害を表し、通常筋原性の症状を指す。パールカンは神経筋接合部の基底膜にアセチルコリンエステラーゼを結合させ、アセチルコリンによる神経筋の興奮とその解除を調節していることから、パールカンの変異や欠損により筋の収縮活動に異常をきたすと考えられている (39)。
シルバーマン・ハンドマーカー型分節異常骨異形成症の臨床症状の特徴は、四肢が短く、不同等の椎体骨、扁平顔、小顎症、口蓋裂、関節可動域の低下、脳ヘルニアなどを示し周産期に死亡する (36, 37)。一方、ローランド・デビュクオア型分節異常骨異形成症は、シルバーマン・ハンドマーカー型と同様の症状を示すが、致死には至らない (38)。シルバーマン・ハンドマーカー型とローランド・デビュクオア型の致死性か致死性でない違いは、おそらくHSPG2の変異部位に依存しているのであろう (38)。
AGRNによってコードされるアグリンは、2,068アミノ酸から構成され、1分子あたり3箇所のHS付加部位をもつ。神経や神経筋接合部の細胞外マトリックスに多く存在している。AGRNの変異によって、常染色体潜性の神経筋伝達障害である筋無力症候群が発症する (40-43)。本疾患は、神経終末細胞骨格の変化やシナプス間隙の分裂など神経筋接合部の破綻が生じ、結果としてアセチルコリン受容体のクラスター形成の低下、筋特異的チロシンキナーゼ受容体のリン酸化が減少することで、筋力低下が引き起こされる。
COL18A1によってコードされる18α1型コラーゲンは、1,339アミノ酸から構成され、1分子あたり3箇所のHS付加部位をもつ。細胞によってはCSとのハイブリッド型PGの形をとる場合もある。COL18A1の変異によって、常染色体潜性の1型ノブロフ症候群が発症する (44-47)。本疾患の臨床的特徴は、強度近視、網膜剥離を伴う硝子体網膜変性、黄斑部異常、後頭部の脳ヘルニアなどである。さらに、COL18A1の変異で常染色体顕性の原発閉塞隅角緑内障を発症する (48)。本疾患患者の眼は、虹彩の周囲と線維柱帯網が接触しているため、前房隅角が閉鎖することにより、房水の流出が阻害される。その結果、眼圧の上昇を招く。
4. ケラタン硫酸プロテオグリカン欠損症
KERAによってコードされるケラトカンは、352アミノ酸から構成され、1分子あたり3箇所のKS付加部位(アスパラギン残基)をもつ。KERAの変異によって、常染色体潜性の2型扁平角膜症が発症する (49-58)。本疾患の臨床的特徴は、遠視、角膜曲率(カーブの度合い)の低下、角膜縁の濁り、脂質が沈着した角膜環(老人環)などである。
5. おわりに
上述したように、各PGのコアタンパク質の変異や欠損により、骨格・皮膚・血管・眼・神経などの遺伝性疾患が引き起こされる。これらは各GAG (CS/DS/HS/KS) の欠損に基づく遺伝性疾患と共通している部分が多い(CS/DS/HS/KSの生合成機構と遺伝性疾患については、本シリーズの他の項を参照されたい)。一方で、PGのコアタンパク質の違いは、その機能の違いは元より、発現分布の違いによって、各遺伝性疾患の臨床症状は異なっている。今後、患者由来細胞やノックアウトマウスを用いた詳細な解析により、発症メカニズムの解明ならびに治療法・治療薬の開発に繋がることを期待している。
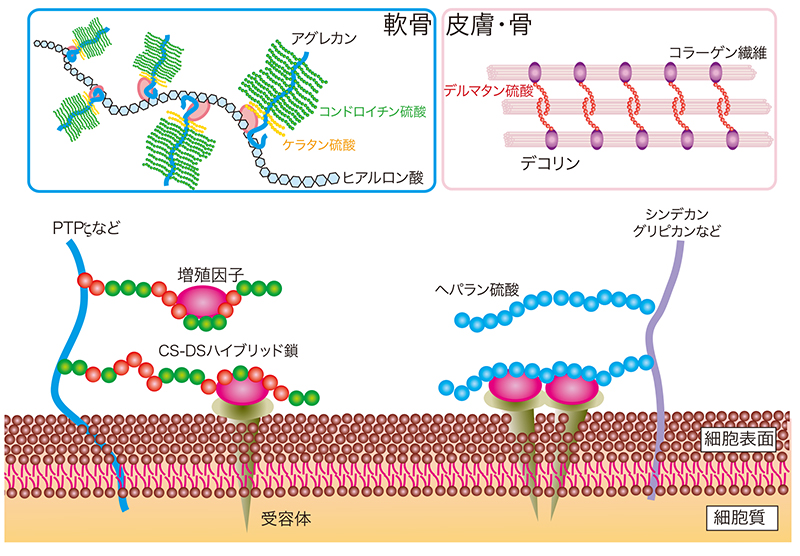
図 1. 細胞表面や細胞外マトリックスに存在するプロテオグリカンの機能
|
表 1. プロテオグリカンの変異または欠損による遺伝病
| 遺伝子 | タンパク質 | 染色体 | MIM*ナンバー | 遺伝病名 |
| ACAN | Aggrecan | 15q26.1 | 155760
165800
608361
612813
| キンバリー型脊椎骨端異形成症; アグリカン型脊椎骨端異形成症; 早発性の変形性関節症または離断性骨軟骨炎を伴う(伴わない)低身長と亢進した骨年齢 |
| VCAN | Versican | 5q14.2-q14.3 | 118661
143200
| ワーグナー症候群1 |
| BGN | Biglycan | Xq28 | 300106
300989
301870
| メーステル・ロイス症候群; X染色体性脊椎骨端骨幹端異形成症 |
| DCN | Decorin | 12q21.33 | 125255
610048
| 角膜実質ジストロフィー |
| GPC3 | Glypican 3 | Xq26.2 | 300037
312870
| 1型シンプソン・ゴラビ・ベーメル症候群 |
| GPC4 | Glypican 4 | Xq26.2 | 300168
301026
| ケイパート症候群 |
| GPC6 | Glypican 6 | 13q31.3-q32.1 | 258315
604404
| 肩異形成症1 |
| HSPG2 | Perlecan | 1p36.12 | 142461
224410
255800
| 1型シュワルツ・ヤンペル症候群; シルバーマン・ハンドマーカー型分節異常骨異形成症; ローランド・デビュクオア分節異常骨異形成症 |
| AGRN | Agrin | 1p36.33 | 103320
615120
| シナプス前後部の欠損を伴う遺伝性筋無力症候群8 |
| COL18A1 | Collagen 18α1 | 21q22.3 | 120328
267750
618880
| 1型ノブロフ症候群; 原発閉塞隅角緑内障 |
KERA | Keratocan | 12q21.33 | 217300
603288
| 常染色体劣性扁平角膜症2 |
* MIM, Mendelian inheritance in man
表 2. プロテオグリカンの変異・欠損に起因する遺伝性疾患に関する参考文献
|
遺伝子 |
原著論文
|
| ACAN |
(7) Gleghorn L, Ramesar R, Beighton P, Wallis G: A mutation in the variable repeat region of the aggrecan gene (AGC1) causes a form of spondyloepiphyseal dysplasia associated with severe, premature osteoarthritis. Am. J. Hum. Genet. 77, 484-490, 2005.
(8) Tompson SW, Merriman B, Funari VA, Fresquet M, Lachman RS, Rimoin DL, Nelson SF, Briggs MD, Cohn DH, Krakow D: A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C-type lectin domain of aggrecan. Am. J. Hum. Genet. 84, 72-79, 2009.
(9) Stattin EL, Wiklund F, Lindblom K, Onnerfjord P, Jonsson BA, Tegner Y, Sasaki T, Struglics A, Lohmander S, Dahl N, Heinegård D, Aspberg A: A missense mutation in the aggrecan C-type lectin domain disrupts extracellular matrix interactions and causes dominant familial osteochondritis dissecans. Am. J. Hum. Genet. 86, 126-137, 2010. |
| VCAN |
(10) Miyamoto T, Inoue H, Sakamoto Y, Kudo E, Naito T, Mikawa T, Mikawa Y, Isashiki Y, Osabe D, Shinohara S, Shiota H, Itakura M: Identification of a novel splice site mutation of the CSPG2 gene in a Japanese family with Wagner syndrome. Invest. Ophthalmol. Vis. Sci. 46, 2726-2735, 2005.
(11) Kloeckener-Gruissem B, Bartholdi D, Abdou MT, Zimmermann DR, Berger W: Identification of the genetic defect in the original Wagner syndrome family. Mol. Vis. 12, 350-355, 2006.
(12) Kloeckener-Gruissem B, Neidhardt J, Magyar I, Plauchu H, Zech JC, Morlé L, Palmer-Smith SM, Macdonald MJ, Nas V, Fry AE, Berger W: Novel VCAN mutations and evidence for unbalanced alternative splicing in the pathogenesis of Wagner syndrome. Eur. J. Hum. Genet. 21, 352-356, 2013.
|
| BGN |
(13) Cho SY, Bae JS, Kim NKD, Forzano F, Girisha KM, Baldo C, Faravelli F, Cho TJ, Kim D, Lee KY, Ikegawa S, Shim JS, Ko AR, Miyake N, Nishimura G, Superti-Furga A, Spranger J, Kim OH, Park WY, Jin DK: BGN mutations in X-linked spondyloepimetaphyseal dysplasia. Am. J. Hum. Genet. 98, 1243-1248, 2016.
(14) Meester JA, Vandeweyer G, Pintelon I, Lammens M, Van Hoorick L, De Belder S, Waitzman K, Young L, Markham LW, Vogt J, Richer J, Beauchesne LM, Unger S, Superti-Furga A, Prsa M, Dhillon R, Reyniers E, Dietz HC, Wuyts W, Mortier G, Verstraeten A, Van Laer L, Loeys BL: Loss-of-function mutations in the X-linked biglycan gene cause a severe syndromic form of thoracic aortic aneurysms and dissections. Genet. Med. 19, 386-395, 2017.
|
| DCN
|
(15) Van Ginderdeuren R, De Vos R, Casteels I, Foets B: Report of a new family with dominant congenital heredity stromal dystrophy of the cornea. Cornea 21, 118-120, 2002.
(16) Bredrup C, Knappskog PM, Majewski J, Rødahl E, Boman H: Congenital stromal dystrophy of the cornea caused by a mutation in the decorin gene. Invest. Ophthalmol. Vis. Sci. 46, 420-426, 2005.
(17) Rødahl E, Van Ginderdeuren R, Knappskog PM, Bredrup C, Boman H: A second decorin frame shift mutation in a family with congenital stromal corneal dystrophy. Am. J. Ophthalmol. 142, 520-521, 2006.
(18) Kim JH, Ko JM, Lee I, Kim JY, Kim MJ, Tchah H: A novel mutation of the decorin gene identified in a Korean family with congenital hereditary stromal dystrophy. Cornea 30, 1473-1477, 2011.
(19) Jing Y, Kumar PR, Zhu L, Edward DP, Tao S, Wang L, Chuck R, Zhang C: Novel decorin mutation in a Chinese family with congenital stromal corneal dystrophy. Cornea 33, 288-293, 2014.
(20) Williams D, Chung DD, Hovakimyan A, Davtyan A, Glasgow BJ, Aldave AJ: Novel DCN mutation in Armenian family with congenital stromal corneal dystrophy. Cornea 42, 464-469, 2023. |
| GPC3 |
(21) Veugelers M, Cat BD, Muyldermans SY, Reekmans G, Delande N, Frints S, Legius E, Fryns JP, Schrander-Stumpel C, Weidle B, Magdalena N, David G: Mutational analysis of the GPC3/GPC4 glypican gene cluster on Xq26 in patients with Simpson-Golabi-Behmel syndrome: identification of loss-of-function mutations in the GPC3 gene. Hum. Mol. Genet. 9, 1321-1328, 2000.
(22) Sakazume S, Okamoto N, Yamamoto T, Kurosawa K, Numabe H, Ohashi Y, Kako Y, Nagai T, Ohashi H: GPC3 mutations in seven patients with Simpson-Golabi-Behmel syndrome. Am. J. Med. Genet. A 143A, 1703-1707, 2007.
(23) Vuillaume ML, Moizard MP, Rossignol S, Cottereau E, Vonwill S, Alessandri JL, Busa T, Colin E, Gérard M, Giuliano F, Lambert L, Lefevre M, Kotecha U, Nampoothiri S, Netchine I, Raynaud M, Brioude F, Toutain A: Mutation update for the GPC3 gene involved in Simpson-Golabi-Behmel syndrome and review of the literature. Hum. Mutat. 39, 790-805, 2018.
(24) Fernandes C, Paúl A, Venâncio MM, Ramos F: Simpson-Golabi-Behmel syndrome: One family, same mutation, different outcome. Am. J. Med. Genet. A. 185, 2502-2506, 2021.
(25) Watanabe K, Noguchi A, Takahashi I, Yamada M, Suzuki H, Takenouchi T, Kosaki K, Takahashi T: Precocious puberty in a case of Simpson-Golabi-Behmel syndrome with a de novo 240-kb deletion including GPC3. Hum. Genome Var. 9, 23, 2022.
|
| GPC4 |
(26) Amor DJ, Stephenson SEM, Mustapha M, Mensah MA, Ockeloen CW, Lee WS, Tankard RM, Phelan DG, Shinawi M, de Brouwer APM, Pfundt R, Dowling C, Toler TL, Sutton VR, Agolini E, Rinelli M, Capolino R, Martinelli D, Zampino G, Dumić M, Reardon W, Shaw-Smith C, Leventer RJ, Delatycki MB, Kleefstra T, Mundlos S, Mortier G, Bahlo M, Allen NJ, Lockhart PJ: Pathogenic variants in GPC4 cause Keipert syndrome. Am. J. Hum. Genet. 104, 914-924, 2019.
(27) Kuroda Y, Uehara T, Enomoto Y, Naruto T, Matsumura N, Kurosawa K: GPC4 truncating variant associated with Keipert syndrome and lacrimal punctal agenesis. Am. J. Med. Genet. A 194, e63799, 2024. |
| GPC6 |
(28) Campos-Xavier AB, Martinet D, Bateman J, Belluoccio D, Rowley L, Tan TY, Baxová A, Gustavson KH, Borochowitz ZU, Innes AM, Unger S, Beckmann JS, Mittaz L, Ballhausen D, Superti-Furga A, Savarirayan R, Bonafé L: Mutations in the heparan-sulfate proteoglycan glypican 6 (GPC6) impair endochondral ossification and cause recessive omodysplasia. Am. J. Hum. Genet. 84, 760-770, 2009.
(29) Crenshaw MM, Meyers ML, Brown K, Slegesky V, Duis J, Elias ER, Saenz M, Shi W, Filmus J, Meeks NJL: Five siblings expand the spectrum of GPC6-related skeletal dysplasia. Am. J. Med. Genet. A 191, 2571-2577, 2023. |
| HSPG2 |
(30) Nicole S, Davoine CS, Topaloglu H, Cattolico L, Barral D, Beighton P, Hamida CB, Hammouda H, Cruaud C, White PS, Samson D, Urtizberea JA, Lehmann-Horn F, Weissenbach J, Hentati F, Fontaine B: Perlecan, the major proteoglycan of basement membranes, is altered in patients with Schwartz-Jampel syndrome (chondrodystrophic myotonia). Nat. Genet. 26, 480-483, 2000.
(31) Arikawa-Hirasawa E, Le AH, Nishino I, Nonaka I, Ho NC, Francomano CA, Govindraj P, Hassell JR, Devaney JM, Spranger J, Stevenson RE, Iannaccone S, Dalakas MC, Yamada Y: Structural and functional mutations of the perlecan gene cause Schwartz-Jampel syndrome, with myotonic myopathy and chondrodysplasia. Am. J. Hum. Genet. 70, 1368-1375, 2002.
(32) Stum M, Davoine CS, Vicart S, Guillot-Noël L, Topaloglu H, Carod-Artal FJ, Kayserili H, Hentati F, Merlini L, Urtizberea JA, Hammouda el-H, Quan PC, Fontaine B, Nicole S: Spectrum of HSPG2 (Perlecan) mutations in patients with Schwartz-Jampel syndrome. Hum. Mutat. 27, 1082-1091, 2006.
(33) Iwata S, Ito M, Nakata T, Noguchi Y, Okuno T, Ohkawara B, Masuda A, Goto T, Adachi M, Osaka H, Nonaka R, Arikawa-Hirasawa E, Ohno K: A missense mutation in domain III in HSPG2 in Schwartz-Jampel syndrome compromises secretion of perlecan into the extracellular space. Neuromuscul. Disord. 25, 667-671, 2015.
(34) Das Bhowmik A, Dalal A, Matta D, Kandadai RM, Kanikannan MA, Aggarwal S: Identification of a novel splice site HSPG2 mutation and prenatal diagnosis in Schwartz Jampel Syndrome type 1 using whole exome sequencing. Neuromuscul. Disord. 26, 809-814, 2016.
(35) Elahi Vahed I, Tehrani Fateh S, Kamali M, Hashemi-Gorji F, Esmaeilzadeh Z, Sadeghi H, Miryounesi M, Ghasemi MR: Expanding genetic and clinical aspects of Schwartz-Jampel syndrome: A report of two cases with literature review. Mol. Genet. Metab. Rep. 40, 101125, 2024.
(36) Arikawa-Hirasawa E, Wilcox WR, Le AH, Silverman N, Govindraj P, Hassell JR, Yamada Y: Dyssegmental dysplasia, Silverman-Handmaker type, is caused by functional null mutations of the perlecan gene. Nat. Genet. 27, 431-434, 2001.
(37) Basalom S, Trakadis Y, Shear R, Azouz ME, De Bie I: Dyssegmental dysplasia, Silverman-Handmaker type: A challenging antenatal diagnosis in a dizygotic twin pregnancy. Mol. Genet. Genomic Med. 6, 452-456, 2018.
(38) Farshadyeganeh P, Yamada T, Ohashi H, Nishimura G, Fujita H, Oishi Y, Nunode M, Ishikawa S, Murotsuki J, Yamashita Y, Ikegawa S, Ogi T, Arikawa-Hirasawa E, Ohno K: Dyssegmental dysplasia Rolland-Desbuquois type is caused by pathogenic variants in HSPG2 - a founder haplotype shared in five patients. J. Hum. Genet. 69, 235-244, 2024.
(39) Arikawa-Hirasawa E: Impact of the heparan sulfate proteoglycan perlecan on human disease and health. Am. J. Physiol. Cell Physiol. 322, C1117-C1122, 2022. |
| AGRN |
(40) Huzé C, Bauché S, Richard P, Chevessier F, Goillot E, Gaudon K, Ben Ammar A, Chaboud A, Grosjean I, Lecuyer HA, Bernard V, Rouche A, Alexandri N, Kuntzer T, Fardeau M, Fournier E, Brancaccio A, Rüegg MA, Koenig J, Eymard B, Schaeffer L, Hantaï D: Identification of an agrin mutation that causes congenital myasthenia and affects synapse function. Am. J. Hum. Genet. 85, 155-167, 2009.
(41) Maselli RA, Fernandez JM, Arredondo J, Navarro C, Ngo M, Beeson D, Cagney O, Williams DC, Wollmann RL, Yarov-Yarovoy V, Ferns MJ: LG2 agrin mutation causing severe congenital myasthenic syndrome mimics functional characteristics of non-neural (z-) agrin. Hum Genet. 131, 1123-11235, 2012.
(42) Ohkawara B, Shen X, Selcen D, Nazim M, Bril V, Tarnopolsky MA, Brady L, Fukami S, Amato AA, Yis U, Ohno K, Engel AG: Congenital myasthenic syndrome-associated agrin variants affect clustering of acetylcholine receptors in a domain-specific manner. JCI Insight 5, e132023, 2020.
(43) Gharebaghian H, Ghasemi A, Omid Hesami, Nafissi S: Identifying novel AGRN variants in congenital myasthenic syndrome: insights from three Iranian families. Neuromuscul. Disord. 49, 105342, 2025. |
| COL18A1 |
(44) Sertié AL, Sossi V, Camargo AA, Zatz M, Brahe C, Passos-Bueno MR: Collagen XVIII, containing an endogenous inhibitor of angiogenesis and tumor growth, plays a critical role in the maintenance of retinal structure and in neural tube closure (Knobloch syndrome). Hum. Mol. Genet. 9, 2051-2058, 2000.
(45) Suzuki OT, Sertié AL, Der Kaloustian VM, Kok F, Carpenter M, Murray J, Czeizel AE, Kliemann SE, Rosemberg S, Monteiro M, Olsen BR, Passos-Bueno MR: Molecular analysis of collagen XVIII reveals novel mutations, presence of a third isoform, and possible genetic heterogeneity in Knobloch syndrome. Am. J. Hum. Genet. 71, 1320-1329, 2002.
(46) Caglayan AO, Baranoski JF, Aktar F, Han W, Tuysuz B, Guzel A, Guclu B, Kaymakcalan H, Aktekin B, Akgumus GT, Murray PB, Erson-Omay EZ, Caglar C, Bakircioglu M, Sakalar YB, Guzel E, Demir N, Tuncer O, Senturk S, Ekici B, Minja FJ, Šestan N, Yasuno K, Bilguvar K, Caksen H, Gunel M: Brain malformations associated with Knobloch syndrome--review of literature, expanding clinical spectrum, and identification of novel mutations. Pediatr. Neurol. 51, 806-813, 2014.
(47) Corbett MA, Turner SJ, Gardner A, Silver J, Stankovich J, Leventer RJ, Derry CP, Carroll R, Ha T, Scheffer IE, Bahlo M, Jackson GD, Mackey DA, Berkovic SF, Gecz J: Familial epilepsy with anterior polymicrogyria as a presentation of COL18A1 mutations. Eur. J. Med. Genet. 60, 437-443, 2017.
(48) Suri F, Yazdani S, Chapi M, Safari I, Rasooli P, Daftarian N, Jafarinasab MR, Ghasemi Firouzabadi S, Alehabib E, Darvish H, Klotzle B, Fan JB, Turk C, Elahi E: COL18A1 is a candidate eye iridocorneal angle-closure gene in humans. Hum. Mol. Genet. 27, 3772-3786, 2018.
|
| KERA |
(49) Tahvanainen E, Forsius H, Kolehmainen J, Damsten M, Fellman J, de la Chapelle A: The genetics of cornea plana congenita. J. Med. Genet. 33, 116-119, 1996.
(50) Pellegata NS, Dieguez-Lucena JL, Joensuu T, Lau S, Montgomery KT, Krahe R, Kivelä T, Kucherlapati R, Forsius, H., and de la Chapelle, A: Mutations in KERA, encoding keratocan, cause cornea plana. Nat. Genet. 25, 91-95, 2000.
(51) Lehmann OJ, El-ashry MF, Ebenezer ND, Ocaka L, Francis PJ, Wilkie SE, Patel RJ, Ficker L, Jordan T, Khaw PT, Bhattacharya SS: A novel keratocan mutation causing autosomal recessive cornea plana. Invest. Ophthalmol. Vis. Sci. 42, 3118-3122, 2001.
(52) Khan A, Al-Saif A, Kambouris M: A novel KERA mutation associated with autosomal recessive cornea plana. Ophthalmic Genet. 25, 147-152, 2004.
(53) Ebenezer ND, Patel CB, Hariprasad SM, Chen LL, Patel RJ, Hardcastle AJ, Allen RC: Clinical and molecular characterization of a family with autosomal recessive cornea plana. Arch. Ophthalmol. 123, 1248-1253, 2005.
(54) Khan AO, Aldahmesh M, Al-Saif A, Meyer B: Pellucid marginal degeneration coexistent with cornea plana in one member of a family exhibiting a novel KERA mutation. Br. J. Ophthalmol. 89, 1538-1540, 2005.
(55) Liskova P, Hysi PG, Williams D, Ainsworth JR, Shah S, de la Chapelle A, Tuft SJ, Bhattacharya SS: Study of p.N247S KERA mutation in a British family with cornea plana. Mol. Vis. 13, 1339-1347, 2007.
(56) Roos L, Bertelsen B, Harris P, Bygum A, Jensen H, Grønskov K, Tümer Z: Case report: a novel KERA mutation associated with cornea plana and its predicted effect on protein function. BMC Med. Genet. 16, 40, 2015.
(57) Kumari D, Tiwari A, Choudhury M, Kumar A, Rao A, Dixit M: A novel KERA mutation in a case of autosomal recessive cornea plana with primary angle-closure glaucoma. J. Glaucoma 25, e106-109, 2016.
(58) Dudakova L, Vercruyssen JHJ, Balikova I, Postolache L, Leroy BP, Skalicka P, Liskova P: Analysis of KERA in four families with cornea plana identifies two novel mutations. Acta Ophthalmol. 96, e87-e91, 2018. |
水本 秀二(名城大学薬学部病態生化学研究室)
| References |
| (1) |
Kjellén L, Lindahl U: Proteoglycans: structures and interactions. Annu. Rev. Biochem. 60, 443-475, 1991 |
| (2) |
Iozzo RV: Matrix proteoglycans: from molecular design to cellular function. Annu. Rev. Biochem. 67, 609-652, 1998 |
| (3) |
Bishop JR, Schuksz M, Esko JD: Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446, 1030-1037, 2007 |
| (4) |
Mizumoto S, Yamada S, Sugahara K: Molecular interactions between chondroitin-dermatan sulfate and growth factors/receptors/matrix proteins. Curr. Opin. Struct. Biol. 34, 35-42, 2015 |
| (5) |
Gomez Toledo A, Golden GJ, Cummings RD, Malmström J, Esko JD: Endothelial glycocalyx turnover in vascular health and disease: rethinking endothelial dysfunction. Annu. Rev. Biochem. 94, Online ahead of print, 2025, doi: 10.1146/annurev-biochem-032620-104745. |
| (6) |
Mizumoto S, Yamada S: Congenital disorders of deficiency in glycosaminoglycan biosynthesis. Front. Genet. 12, 717535, 2021 |
2025年 7月17日
|
|---|





