 |
Proteoglycans (PGs) contain glycosaminoglycan (GAG) side chains, and play roles in the regulation of signal transductions on cell surface and assembly of the extracellular matrix (Figure 1) (1-5). GAGs forming proteoglycans include chondroitin sulfate (CS), dermatan sulfate (DS), heparan sulfate (HS), and keratan sulfate (KS), classified as CS-PG, DS-PG, HS-PG, and KS-PG, respectively. CS, DS, HS, and KS are abundant in cartilage, skin and the aorta, liver and kidney, and cornea and cartilage, respectively. The pathogenic variants of genes encoding core protein of PGs cause skeletal and neurological anormalies, heart defects, and eye and ear disorders (Table 1) (6). Details of biosynthetic mechanisms and congenital disorders of CS/DS/HS/KS are referred to in other sections in this series.
1. Defects in chondroitin sulfate-proteoglycans
Aggrecan encoded by ACAN is a protein with 2,431 amino acids modified by CS (> 100 chains per monomer), KS, and N-glycans. Pathogenic variants of ACAN cause autosomal dominant spondyloepiphyseal dysplasia Kimberley/aggrecan types (7-9). Clinical manifestations are characterized by a short stature, stocky build, early-onset osteoarthritis of joints, flattened vertebral bodies with sclerosis and end-plate irregularity, flattened femoral epiphyses, and osteochondritis dissecans.
Versican encoded by VCAN is comprised of a protein with 3,396 amino acids, which has ~20 putative CS attachment sites as well as potential O- and N-glycosylation sites. Pathogenic variants of VCAN cause autosomal dominant Wagner syndrome, characterized by vitreoretinopathy, an empty vitreous cavity, vitreous degeneration, progressive chorioretinal atrophy, perivascular sheathing, subcapsular cataract, and myopia (10-12).
2. Defects in dermatan sulfate-proteoglycans
Biglycan encoded by BGN is a protein with 368 amino acids containing 2 putative DS attachment sites. Pathogenic variants of BGN cause the X-linked form of spondyloepimetaphyseal dysplasia, characterized by anomalies of the spine as well as the epi- and metaphyses of long bones, thereby resulting in a short stature and osteoarthritic changes in joints (13). Furthermore, Meester-Loeys syndrome (thoracic aortic aneurysms and dissections) is also caused by mutations in BGN (14). Clinical manifestations of Meester-Loeys syndrome are characterized by early-onset aortic aneurysm and dissection, joint hypermobility and contractures, deformities of the skin striae and pectus, a bifid uvula and cervical spine instability, and craniofacial dysmorphisms including dolichocephaly, hypertelorism, down-slanting eyes, a high-arched plate, proptosis, malar hypoplasia, and frontal bossing. These clinical features of Meester-Loeys syndrome are similar to those of Loeys-Dietz syndrome, Marfan syndrome, and vascular type Ehlers-Danlos syndrome, caused by mutations in transforming growth factor-β (TGF-β)/TGF-β receptor, fibrillin-1 (FBN1), and type III collagen, respectively, suggesting that biglycan and/or its side chain, DS, contributes to the regulation of TGF-β signaling and assembly of collagen fiber bundles. In fact, biglycan binds to TGF-β in vitro, and collagen fibrils are bundled by interaction with DS.
Decorin encoded by DCN is comprised of a protein with 359 amino acids containing a DS attachment site. Pathogenic variants of DCN cause autosomal dominant congenital stromal corneal dystrophy (15-20). Interestingly, all patients reported to date have had the truncated form of decorin, and no single amino acid substitution. The truncated form of decorin may function as a dominant-negative mutant. Congenital stromal corneal dystrophy is characterized by corneal clouding with fine opacities observed as small flakes and spots, refractive errors, amblyopia, strabismus, nystagmus, and esotropia.
3. Defects in heparan sulfate-proteoglycans
Six members of glypican family proteins are known in humans, and glypicans 1-6 encoded by GPC1 to GPC6 fall into two broad subfamilies: glypicans 1/2/4/6 and glypicans 3/5, with approximately 25% amino-acid identity between groups. Glypican family proteins are made up of 550-580 amino acids containing two putative HS attachment sites at the C-terminus, and are anchored to the plasma membrane by a glycosylphosphatidylinositol (GPI) anchor. Pathogenic variants of GPC3 cause X-linked Simpson-Golabi-Behmel syndrome type 1, characterized by pre- and postnatal overgrowth, visceral and skeletal anormalies, a coarse face, heart defects, and hypotonia (21-25). Pathogenic variants of GPC4 cause Keipert syndrome, known as naso-digito-acoustic syndrome, characterized by craniofacial and digital abnormalities, variable learning difficulties, and sensorineural deafness (26, 27). Pathogenic variants of GPC6 cause autosomal recessive omodysplasia, characterized by proximally shortened limbs, facial dysmorphism, a severely short stature, and hypoplastic humeri (28, 29).
Perlecan encoded by HSPG2 is comprised of a protein with 4,392 amino acids, which is a major HS-PG with two or three HS as well as one or two CS. Perlecan is distributed to the basement membrane as well as extracellular matrix of muscle, cartilage, and bone marrow. Ppathogenic variants of HSPG2 cause autosomal recessive Schwartz-Jampel syndrome type 1/chondrodystrophic myotonia and the Dyssegmental dysplasia Silverman-Handmaker type or Rolland-Desbuquois type (30-38).
Clinical manifestations of Schwartz-Jampel syndrome type 1 are permanent myotonia including prolonged failure of muscle relaxation and skeletal dysplasia including a short stature, kyphoscoliosis, bowing of the diaphysis, and irregular epiphyses (30-35). Myotonia is a myogenic symptom such as impaired muscle with sustained contraction and relaxation. Perlecan binds acetylcholinesterase to the basement membrane of the neuromuscular junction, and regulates the excitation and release of neuromuscular signals by acetylcholine. Therefore, mutations or deficiencies of perlecan cause abnormalities in muscle contraction (39).
Clinical manifestations of Dyssegmental dysplasia Silverman-Handmaker type are skeletal dysplasia with micromelia, anisospondyly, a flat face, micrognathia, cleft palate, reduced joint mobility, encephalocoele, and perinatal death (36, 37). In contrast, clinical manifestations of Dyssegmental dysplasia Rolland-Desbuquois type are similar to those of Silverman-Handmaker type, but not lethal (38). The difference in lethality being present or absent between Silverman-Handmaker and Rolland-Desbuquois type, respectively, may be dependent on the site of HSPG2 mutations (38).
Agrin encoded by AGRN consists of a protein with 2,068 amino acids containing three potential HS attachments sites and an extracellular matrix molecule released by nerves and critical for the formation of neuromuscular junctions. Pathogenic variants of AGRN cause an autosomal recessive neuromuscular transmission disorder, myasthenic syndrome (40-43), characterized by major disorganizations of neuromuscular junctions, including alterations in the nerve-terminal cytoskeleton and fragmentation of the synaptic gutters, resulting in decreased clustering of acetylcholine receptors and decreased phosphorylation of muscle-specific tyrosine kinase receptors, thereby leading to muscle weakness.
Collagen18α1 encoded by COL18A1 consists of a protein with 1,339 amino acids containing three potential HS attachments sites, or the CS/HS hybrid form, which may be dependent on distinct cell types or tissues. Pathogenic variants of COL18A1 cause autosomal recessive Knobloch syndrome type 1, characterized by the occurrence of high myopia, vitreoretinal degeneration with retinal detachment, macular abnormalities, and occipital encephalocele (44-47). Furthermore, pathogenic variants of COL18A1 also cause autosomal dominant primary angle-closure glaucoma (48). The eyes of patients show a closed-anterior chamber angle due to contact between the peripheral iris and posterior trabecular meshwork, which obstructs the outflow of aqueous humor, leading to increased intraocular pressure.
4. Defects in keratan sulfate-proteoglycans
Keratocan encoded by KERA comprises of a protein core of 352 amino acids with three covalent KS attachment sites at asparagine residues. Pathogenic variants of KERA cause autosomal recessive cornea plana type 2, characterized by hyperopia, a reduced corneal curvature, hazy corneal limbus, and arcus lipoides (49-58).
5. Conclusions and Perspectives
As described above, mutations or defects in core proteins of each PG cause genetic disorders of the skeleton, skin, blood vessels, eyes, and nerves. These disorders are common to genetic diseases caused by defects in each GAG (CS/DS/HS/KS) (for biosynthetic mechanisms and genetic disorders of CS/DS/HS/KS, please refer to other sections in this series). However, differences in the core proteins of PGs as well as their functions lead to distinct clinical manifestations of each genetic disorder. Detailed analyses using patients’ cells and knockout mice may lead to elucidation of the onset mechanism and development of treatments and drugs.
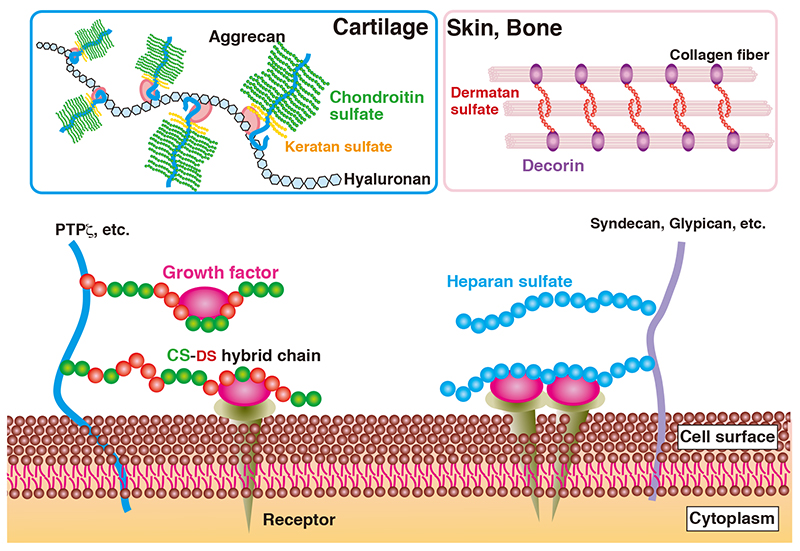
Figure 1. Roles of proteoglycans in the extracellular matrix and on the cell surface
|
Table 1. Congenital disorders of proteoglycan-deficiency
| Gene | Protein | Chromosomal location | MIM* number | Human genetic disorder |
| ACAN | Aggrecan | 15q26.1 | 155760
165800
608361
612813
| Spondyloepiphyseal dysplasia Kimberley type; Spondyloepimetaphyseal dysplasia, aggrecan type; Short stature and advanced bone age, with or without early-onset osteoarthritis and/or osteochondritis dissecans |
| VCAN | Versican | 5q14.2-q14.3 | 118661
143200
| Wanger syndrome 1 |
| BGN | Biglycan | Xq28 | 300106
300989
301870
| Meester-Loeys syndrome; Spondyloepimetaphyseal dysplasia, X-linked |
| DCN | Decorin | 12q21.33 | 125255
610048
| Corneal dystrophy, congenital stromal |
| GPC3 | Glypican 3 | Xq26.2 | 300037
312870
| Simpson-Golabi-Behmel syndrome type 1 |
| GPC4 | Glypican 4 | Xq26.2 | 300168
301026
| Keipert syndrome |
| GPC6 | Glypican 6 | 13q31.3-q32.1 | 258315
604404
| Omodysplasia 1 |
| HSPG2 | Perlecan | 1p36.12 | 142461
224410
255800
| Schwartz-Jampel syndrome, type 1;
Dyssegmental dysplasia, Silverman-Handmaker type; Rolland–Desbuquois type
|
| AGRN | Agrin | 1p36.33 | 103320
615120
| Myasthenic syndrome, congenital, 8, with pre- and postsynaptic defects |
| COL18A1 | Collagen 18α1 | 21q22.3 | 120328
267750
618880
| Knobloch syndrome type 1; Glaucoma, primary closed-angle |
KERA | Keratocan | 12q21.33 | 217300
603288
| Cornea plana 2, autosomal recessive |
* MIM, Mendelian inheritance in man
Table 2. References for congenital disorders of proteoglycan-deficiency
|
Gene |
Original papers
|
| ACAN |
(7) Gleghorn L, Ramesar R, Beighton P, Wallis G: A mutation in the variable repeat region of the aggrecan gene (AGC1) causes a form of spondyloepiphyseal dysplasia associated with severe, premature osteoarthritis. Am. J. Hum. Genet. 77, 484-490, 2005.
(8) Tompson SW, Merriman B, Funari VA, Fresquet M, Lachman RS, Rimoin DL, Nelson SF, Briggs MD, Cohn DH, Krakow D: A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C-type lectin domain of aggrecan. Am. J. Hum. Genet. 84, 72-79, 2009.
(9) Stattin EL, Wiklund F, Lindblom K, Onnerfjord P, Jonsson BA, Tegner Y, Sasaki T, Struglics A, Lohmander S, Dahl N, Heinegård D, Aspberg A: A missense mutation in the aggrecan C-type lectin domain disrupts extracellular matrix interactions and causes dominant familial osteochondritis dissecans. Am. J. Hum. Genet. 86, 126-137, 2010. |
| VCAN |
(10) Miyamoto T, Inoue H, Sakamoto Y, Kudo E, Naito T, Mikawa T, Mikawa Y, Isashiki Y, Osabe D, Shinohara S, Shiota H, Itakura M: Identification of a novel splice site mutation of the CSPG2 gene in a Japanese family with Wagner syndrome. Invest. Ophthalmol. Vis. Sci. 46, 2726-2735, 2005.
(11) Kloeckener-Gruissem B, Bartholdi D, Abdou MT, Zimmermann DR, Berger W: Identification of the genetic defect in the original Wagner syndrome family. Mol. Vis. 12, 350-355, 2006.
(12) Kloeckener-Gruissem B, Neidhardt J, Magyar I, Plauchu H, Zech JC, Morlé L, Palmer-Smith SM, Macdonald MJ, Nas V, Fry AE, Berger W: Novel VCAN mutations and evidence for unbalanced alternative splicing in the pathogenesis of Wagner syndrome. Eur. J. Hum. Genet. 21, 352-356, 2013.
|
| BGN |
(13) Cho SY, Bae JS, Kim NKD, Forzano F, Girisha KM, Baldo C, Faravelli F, Cho TJ, Kim D, Lee KY, Ikegawa S, Shim JS, Ko AR, Miyake N, Nishimura G, Superti-Furga A, Spranger J, Kim OH, Park WY, Jin DK: BGN mutations in X-linked spondyloepimetaphyseal dysplasia. Am. J. Hum. Genet. 98, 1243-1248, 2016.
(14) Meester JA, Vandeweyer G, Pintelon I, Lammens M, Van Hoorick L, De Belder S, Waitzman K, Young L, Markham LW, Vogt J, Richer J, Beauchesne LM, Unger S, Superti-Furga A, Prsa M, Dhillon R, Reyniers E, Dietz HC, Wuyts W, Mortier G, Verstraeten A, Van Laer L, Loeys BL: Loss-of-function mutations in the X-linked biglycan gene cause a severe syndromic form of thoracic aortic aneurysms and dissections. Genet. Med. 19, 386-395, 2017.
|
| DCN
|
(15) Van Ginderdeuren R, De Vos R, Casteels I, Foets B: Report of a new family with dominant congenital heredity stromal dystrophy of the cornea. Cornea 21, 118-120, 2002.
(16) Bredrup C, Knappskog PM, Majewski J, Rødahl E, Boman H: Congenital stromal dystrophy of the cornea caused by a mutation in the decorin gene. Invest. Ophthalmol. Vis. Sci. 46, 420-426, 2005.
(17) Rødahl E, Van Ginderdeuren R, Knappskog PM, Bredrup C, Boman H: A second decorin frame shift mutation in a family with congenital stromal corneal dystrophy. Am. J. Ophthalmol. 142, 520-521, 2006.
(18) Kim JH, Ko JM, Lee I, Kim JY, Kim MJ, Tchah H: A novel mutation of the decorin gene identified in a Korean family with congenital hereditary stromal dystrophy. Cornea 30, 1473-1477, 2011.
(19) Jing Y, Kumar PR, Zhu L, Edward DP, Tao S, Wang L, Chuck R, Zhang C: Novel decorin mutation in a Chinese family with congenital stromal corneal dystrophy. Cornea 33, 288-293, 2014.
(20) Williams D, Chung DD, Hovakimyan A, Davtyan A, Glasgow BJ, Aldave AJ: Novel DCN mutation in Armenian family with congenital stromal corneal dystrophy. Cornea 42, 464-469, 2023. |
| GPC3 |
(21) Veugelers M, Cat BD, Muyldermans SY, Reekmans G, Delande N, Frints S, Legius E, Fryns JP, Schrander-Stumpel C, Weidle B, Magdalena N, David G: Mutational analysis of the GPC3/GPC4 glypican gene cluster on Xq26 in patients with Simpson-Golabi-Behmel syndrome: identification of loss-of-function mutations in the GPC3 gene. Hum. Mol. Genet. 9, 1321-1328, 2000.
(22) Sakazume S, Okamoto N, Yamamoto T, Kurosawa K, Numabe H, Ohashi Y, Kako Y, Nagai T, Ohashi H: GPC3 mutations in seven patients with Simpson-Golabi-Behmel syndrome. Am. J. Med. Genet. A 143A, 1703-1707, 2007.
(23) Vuillaume ML, Moizard MP, Rossignol S, Cottereau E, Vonwill S, Alessandri JL, Busa T, Colin E, Gérard M, Giuliano F, Lambert L, Lefevre M, Kotecha U, Nampoothiri S, Netchine I, Raynaud M, Brioude F, Toutain A: Mutation update for the GPC3 gene involved in Simpson-Golabi-Behmel syndrome and review of the literature. Hum. Mutat. 39, 790-805, 2018.
(24) Fernandes C, Paúl A, Venâncio MM, Ramos F: Simpson-Golabi-Behmel syndrome: One family, same mutation, different outcome. Am. J. Med. Genet. A. 185, 2502-2506, 2021.
(25) Watanabe K, Noguchi A, Takahashi I, Yamada M, Suzuki H, Takenouchi T, Kosaki K, Takahashi T: Precocious puberty in a case of Simpson-Golabi-Behmel syndrome with a de novo 240-kb deletion including GPC3. Hum. Genome Var. 9, 23, 2022.
|
| GPC4 |
(26) Amor DJ, Stephenson SEM, Mustapha M, Mensah MA, Ockeloen CW, Lee WS, Tankard RM, Phelan DG, Shinawi M, de Brouwer APM, Pfundt R, Dowling C, Toler TL, Sutton VR, Agolini E, Rinelli M, Capolino R, Martinelli D, Zampino G, Dumić M, Reardon W, Shaw-Smith C, Leventer RJ, Delatycki MB, Kleefstra T, Mundlos S, Mortier G, Bahlo M, Allen NJ, Lockhart PJ: Pathogenic variants in GPC4 cause Keipert syndrome. Am. J. Hum. Genet. 104, 914-924, 2019.
(27) Kuroda Y, Uehara T, Enomoto Y, Naruto T, Matsumura N, Kurosawa K: GPC4 truncating variant associated with Keipert syndrome and lacrimal punctal agenesis. Am. J. Med. Genet. A 194, e63799, 2024. |
| GPC6 |
(28) Campos-Xavier AB, Martinet D, Bateman J, Belluoccio D, Rowley L, Tan TY, Baxová A, Gustavson KH, Borochowitz ZU, Innes AM, Unger S, Beckmann JS, Mittaz L, Ballhausen D, Superti-Furga A, Savarirayan R, Bonafé L: Mutations in the heparan-sulfate proteoglycan glypican 6 (GPC6) impair endochondral ossification and cause recessive omodysplasia. Am. J. Hum. Genet. 84, 760-770, 2009.
(29) Crenshaw MM, Meyers ML, Brown K, Slegesky V, Duis J, Elias ER, Saenz M, Shi W, Filmus J, Meeks NJL: Five siblings expand the spectrum of GPC6-related skeletal dysplasia. Am. J. Med. Genet. A 191, 2571-2577, 2023. |
| HSPG2 |
(30) Nicole S, Davoine CS, Topaloglu H, Cattolico L, Barral D, Beighton P, Hamida CB, Hammouda H, Cruaud C, White PS, Samson D, Urtizberea JA, Lehmann-Horn F, Weissenbach J, Hentati F, Fontaine B: Perlecan, the major proteoglycan of basement membranes, is altered in patients with Schwartz-Jampel syndrome (chondrodystrophic myotonia). Nat. Genet. 26, 480-483, 2000.
(31) Arikawa-Hirasawa E, Le AH, Nishino I, Nonaka I, Ho NC, Francomano CA, Govindraj P, Hassell JR, Devaney JM, Spranger J, Stevenson RE, Iannaccone S, Dalakas MC, Yamada Y: Structural and functional mutations of the perlecan gene cause Schwartz-Jampel syndrome, with myotonic myopathy and chondrodysplasia. Am. J. Hum. Genet. 70, 1368-1375, 2002.
(32) Stum M, Davoine CS, Vicart S, Guillot-Noël L, Topaloglu H, Carod-Artal FJ, Kayserili H, Hentati F, Merlini L, Urtizberea JA, Hammouda el-H, Quan PC, Fontaine B, Nicole S: Spectrum of HSPG2 (Perlecan) mutations in patients with Schwartz-Jampel syndrome. Hum. Mutat. 27, 1082-1091, 2006.
(33) Iwata S, Ito M, Nakata T, Noguchi Y, Okuno T, Ohkawara B, Masuda A, Goto T, Adachi M, Osaka H, Nonaka R, Arikawa-Hirasawa E, Ohno K: A missense mutation in domain III in HSPG2 in Schwartz-Jampel syndrome compromises secretion of perlecan into the extracellular space. Neuromuscul. Disord. 25, 667-671, 2015.
(34) Das Bhowmik A, Dalal A, Matta D, Kandadai RM, Kanikannan MA, Aggarwal S: Identification of a novel splice site HSPG2 mutation and prenatal diagnosis in Schwartz Jampel Syndrome type 1 using whole exome sequencing. Neuromuscul. Disord. 26, 809-814, 2016.
(35) Elahi Vahed I, Tehrani Fateh S, Kamali M, Hashemi-Gorji F, Esmaeilzadeh Z, Sadeghi H, Miryounesi M, Ghasemi MR: Expanding genetic and clinical aspects of Schwartz-Jampel syndrome: A report of two cases with literature review. Mol. Genet. Metab. Rep. 40, 101125, 2024.
(36) Arikawa-Hirasawa E, Wilcox WR, Le AH, Silverman N, Govindraj P, Hassell JR, Yamada Y: Dyssegmental dysplasia, Silverman-Handmaker type, is caused by functional null mutations of the perlecan gene. Nat. Genet. 27, 431-434, 2001.
(37) Basalom S, Trakadis Y, Shear R, Azouz ME, De Bie I: Dyssegmental dysplasia, Silverman-Handmaker type: A challenging antenatal diagnosis in a dizygotic twin pregnancy. Mol. Genet. Genomic Med. 6, 452-456, 2018.
(38) Farshadyeganeh P, Yamada T, Ohashi H, Nishimura G, Fujita H, Oishi Y, Nunode M, Ishikawa S, Murotsuki J, Yamashita Y, Ikegawa S, Ogi T, Arikawa-Hirasawa E, Ohno K: Dyssegmental dysplasia Rolland-Desbuquois type is caused by pathogenic variants in HSPG2 - a founder haplotype shared in five patients. J. Hum. Genet. 69, 235-244, 2024.
(39) Arikawa-Hirasawa E: Impact of the heparan sulfate proteoglycan perlecan on human disease and health. Am. J. Physiol. Cell Physiol. 322, C1117-C1122, 2022. |
| AGRN |
(40) Huzé C, Bauché S, Richard P, Chevessier F, Goillot E, Gaudon K, Ben Ammar A, Chaboud A, Grosjean I, Lecuyer HA, Bernard V, Rouche A, Alexandri N, Kuntzer T, Fardeau M, Fournier E, Brancaccio A, Rüegg MA, Koenig J, Eymard B, Schaeffer L, Hantaï D: Identification of an agrin mutation that causes congenital myasthenia and affects synapse function. Am. J. Hum. Genet. 85, 155-167, 2009.
(41) Maselli RA, Fernandez JM, Arredondo J, Navarro C, Ngo M, Beeson D, Cagney O, Williams DC, Wollmann RL, Yarov-Yarovoy V, Ferns MJ: LG2 agrin mutation causing severe congenital myasthenic syndrome mimics functional characteristics of non-neural (z-) agrin. Hum Genet. 131, 1123-11235, 2012.
(42) Ohkawara B, Shen X, Selcen D, Nazim M, Bril V, Tarnopolsky MA, Brady L, Fukami S, Amato AA, Yis U, Ohno K, Engel AG: Congenital myasthenic syndrome-associated agrin variants affect clustering of acetylcholine receptors in a domain-specific manner. JCI Insight 5, e132023, 2020.
(43) Gharebaghian H, Ghasemi A, Omid Hesami, Nafissi S: Identifying novel AGRN variants in congenital myasthenic syndrome: insights from three Iranian families. Neuromuscul. Disord. 49, 105342, 2025. |
| COL18A1 |
(44) Sertié AL, Sossi V, Camargo AA, Zatz M, Brahe C, Passos-Bueno MR: Collagen XVIII, containing an endogenous inhibitor of angiogenesis and tumor growth, plays a critical role in the maintenance of retinal structure and in neural tube closure (Knobloch syndrome). Hum. Mol. Genet. 9, 2051-2058, 2000.
(45) Suzuki OT, Sertié AL, Der Kaloustian VM, Kok F, Carpenter M, Murray J, Czeizel AE, Kliemann SE, Rosemberg S, Monteiro M, Olsen BR, Passos-Bueno MR: Molecular analysis of collagen XVIII reveals novel mutations, presence of a third isoform, and possible genetic heterogeneity in Knobloch syndrome. Am. J. Hum. Genet. 71, 1320-1329, 2002.
(46) Caglayan AO, Baranoski JF, Aktar F, Han W, Tuysuz B, Guzel A, Guclu B, Kaymakcalan H, Aktekin B, Akgumus GT, Murray PB, Erson-Omay EZ, Caglar C, Bakircioglu M, Sakalar YB, Guzel E, Demir N, Tuncer O, Senturk S, Ekici B, Minja FJ, Šestan N, Yasuno K, Bilguvar K, Caksen H, Gunel M: Brain malformations associated with Knobloch syndrome--review of literature, expanding clinical spectrum, and identification of novel mutations. Pediatr. Neurol. 51, 806-813, 2014.
(47) Corbett MA, Turner SJ, Gardner A, Silver J, Stankovich J, Leventer RJ, Derry CP, Carroll R, Ha T, Scheffer IE, Bahlo M, Jackson GD, Mackey DA, Berkovic SF, Gecz J: Familial epilepsy with anterior polymicrogyria as a presentation of COL18A1 mutations. Eur. J. Med. Genet. 60, 437-443, 2017.
(48) Suri F, Yazdani S, Chapi M, Safari I, Rasooli P, Daftarian N, Jafarinasab MR, Ghasemi Firouzabadi S, Alehabib E, Darvish H, Klotzle B, Fan JB, Turk C, Elahi E: COL18A1 is a candidate eye iridocorneal angle-closure gene in humans. Hum. Mol. Genet. 27, 3772-3786, 2018.
|
| KERA |
(49) Tahvanainen E, Forsius H, Kolehmainen J, Damsten M, Fellman J, de la Chapelle A: The genetics of cornea plana congenita. J. Med. Genet. 33, 116-119, 1996.
(50) Pellegata NS, Dieguez-Lucena JL, Joensuu T, Lau S, Montgomery KT, Krahe R, Kivelä T, Kucherlapati R, Forsius, H., and de la Chapelle, A: Mutations in KERA, encoding keratocan, cause cornea plana. Nat. Genet. 25, 91-95, 2000.
(51) Lehmann OJ, El-ashry MF, Ebenezer ND, Ocaka L, Francis PJ, Wilkie SE, Patel RJ, Ficker L, Jordan T, Khaw PT, Bhattacharya SS: A novel keratocan mutation causing autosomal recessive cornea plana. Invest. Ophthalmol. Vis. Sci. 42, 3118-3122, 2001.
(52) Khan A, Al-Saif A, Kambouris M: A novel KERA mutation associated with autosomal recessive cornea plana. Ophthalmic Genet. 25, 147-152, 2004.
(53) Ebenezer ND, Patel CB, Hariprasad SM, Chen LL, Patel RJ, Hardcastle AJ, Allen RC: Clinical and molecular characterization of a family with autosomal recessive cornea plana. Arch. Ophthalmol. 123, 1248-1253, 2005.
(54) Khan AO, Aldahmesh M, Al-Saif A, Meyer B: Pellucid marginal degeneration coexistent with cornea plana in one member of a family exhibiting a novel KERA mutation. Br. J. Ophthalmol. 89, 1538-1540, 2005.
(55) Liskova P, Hysi PG, Williams D, Ainsworth JR, Shah S, de la Chapelle A, Tuft SJ, Bhattacharya SS: Study of p.N247S KERA mutation in a British family with cornea plana. Mol. Vis. 13, 1339-1347, 2007.
(56) Roos L, Bertelsen B, Harris P, Bygum A, Jensen H, Grønskov K, Tümer Z: Case report: a novel KERA mutation associated with cornea plana and its predicted effect on protein function. BMC Med. Genet. 16, 40, 2015.
(57) Kumari D, Tiwari A, Choudhury M, Kumar A, Rao A, Dixit M: A novel KERA mutation in a case of autosomal recessive cornea plana with primary angle-closure glaucoma. J. Glaucoma 25, e106-109, 2016.
(58) Dudakova L, Vercruyssen JHJ, Balikova I, Postolache L, Leroy BP, Skalicka P, Liskova P: Analysis of KERA in four families with cornea plana identifies two novel mutations. Acta Ophthalmol. 96, e87-e91, 2018. |
Shuji Mizumoto
(Department of Pathobiochemistry, Faculty of Pharmacy, Meijo University)
| References |
| (1) |
Kjellén L, Lindahl U: Proteoglycans: structures and interactions. Annu. Rev. Biochem. 60, 443-475, 1991 |
| (2) |
Iozzo RV: Matrix proteoglycans: from molecular design to cellular function. Annu. Rev. Biochem. 67, 609-652, 1998 |
| (3) |
Bishop JR, Schuksz M, Esko JD: Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446, 1030-1037, 2007 |
| (4) |
Mizumoto S, Yamada S, Sugahara K: Molecular interactions between chondroitin-dermatan sulfate and growth factors/receptors/matrix proteins. Curr. Opin. Struct. Biol. 34, 35-42, 2015 |
| (5) |
Gomez Toledo A, Golden GJ, Cummings RD, Malmström J, Esko JD: Endothelial glycocalyx turnover in vascular health and disease: rethinking endothelial dysfunction.
Annu. Rev. Biochem. 94, Online ahead of print, 2025, doi: 10.1146/annurev-biochem-032620-104745. |
| (6) |
Mizumoto S, Yamada S: Congenital disorders of deficiency in glycosaminoglycan biosynthesis. Front. Genet. 12, 717535, 2021 |
Jul. 17, 2025
|
|---|





