 |
Musculocontractural Ehlers-Danlos syndrome (mcEDS) is a representative disorder caused by defective enzymes involving dermatan sulfate (DS) biosynthesis. McEDS is an established type of EDS, classified into two subtypes: a subtype caused by pathogenic variants in CHST14 encoding dermatan 4-O-sulfotransferase 1 (D4ST1), named as mcEDS-CHST14 (MIM#601776); a subtype caused by pathogenic variants in DSE encoding dermatan sulfate epimerase 1 (DS-epi1), named as mcEDS-DSE (MIM#615539) (1-3).
McEDS was originally reported as three independent conditions: a rare congenital arthrogryposis syndrome “adducted thumb-clubfoot syndrome” (4), a specific type of EDS “EDS Kosho type” (5) and a subtype of kyphoscoliotic EDS (6). In 2001, the author encountered the first patient with mcEDS-CHST14. She had multisystem features including congenital arthrogryposis syndrome, facial characteristics, progressive skin laxity/fragility and joint laxity/deformity, dislocation, massive subcutaneous hematomas, and motor developmental delay. In 2003, the author met the second patient with similar features as the first patient, and published a report as a possible new syndrome (7). Because consanguinity was noted in parents of the second patient and also in parents in the third patient the author met in 2007, a genomic research was conducted. After narrowing the candidate region through homozygosity mapping using SNP array and haplotype analysis using flanking microsatellite markers, CHST14 was identified as the causative gene for this disorder. Through a glycobiological research, a defective activity of D4ST1 and complete depletion of DS were observed in patients’ skin fibroblasts; in the glycosaminoglycan (GAG) side chains of decorin, a representative DS-containing proteoglycan, DS was completely replaced by chondroitin sulfate (CS) compared to healthy control subjects containing solely DS. Furthermore, a pathological investigation showed impaired assembly of collagen fibrils in the patients. Because decorin has an important role in assembling collagen fibrils, EDS, disorders of abnormal collagen biosynthesis, were recognized to be linked to this subtype, a disorder of impaired DS biosynthesis (5). We encountered additional five Japanese patients; and, through comprehensive review of all reported patients, the disorder was demonstrated to be a clinically independent condition characterized by congenital malformation-related features (e.g., multiple congenital contracture, facial features, visceral and/or ocular malformations) and progressive fragility-related features (e.g., skin hyperextensibility/fragility/bruisability, joint hypermobility/dislocations/foot or spinal deformities, massive subcutaneous hematomas) (8-10). A recently published international collaborative study organized by us found 66 patients from 48 families, demonstrating that facial characteristics were shared by patients beyond their ethnicities (Figure 1A) and that half of the patients experienced dislocations or massive subcutaneous hematomas before the age of six years (Figure 1B) (11). No DS in patients’ urine samples was detected, which suggested that the disorder represents generalized DS depletion and that urinary DS measurement could be a non-invasive marker for the diagnosis of the disorder and for the evaluation of efficacies for future therapeutic approaches (12). An ultrastructural study demonstrated through a cupromeronic blue staining to visualize GAG chains that affected GAG chains were linear, stretching from the outer surface of collagen fibrils to adjacent fibrils; in contrast, those of controls were curved, maintaining close contact with attached collagen fibrils. This structural alteration of GAG side chains of decorin was proposed to result in spatial disorganization of collagen networks; which presumably disrupted the ring-mesh structure of GAG side chains surrounding collagen fibrils (Figure 2A, 2B) (13).
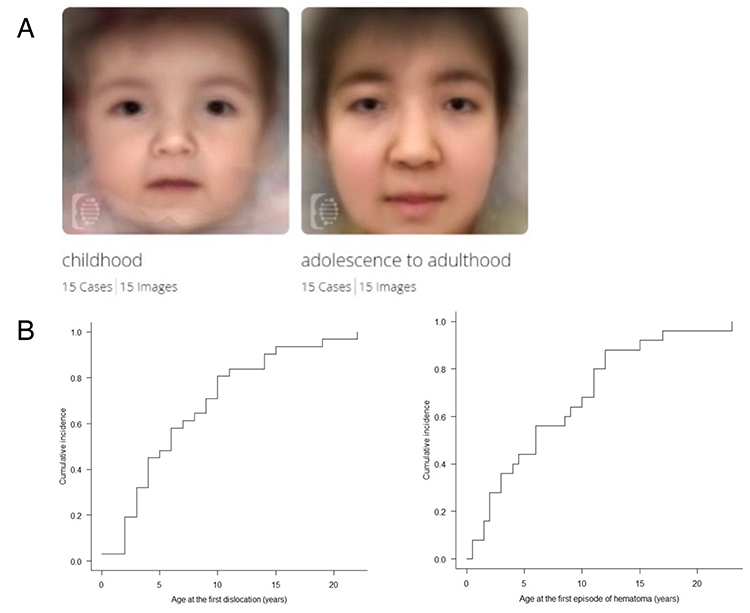
Figure 1. Clinical pictures of mcEDS-CHST14 demonstrated by the international collaborative study (11)
A. Composite images of multiple patients in their childhood patients versus in their adolescence−adulthood using the Face2Gene facial recognition application (https://www.face2gene.com). B. Cumulative incidence of the first episode of dislocation (left) and large cutaneous haematoma (right).
|

Figure 2. Spatial disorganization of skin collagen networks in mcEDS-CHST14 (13)
A. Ultrastructural findings using cupromeronic blue staining of skin specimens (C1-a, a cross-sectional view of a health control subject; P1, P2-a, P3, cross-sectional views of patients; C1-b, a sagittal view of a health control subject; P2-b, a sagittal view of a patient). B. Schematic diagrams of the skin collagen network (a, c, a health control subject; b, d, a patient).
|
Since the first patient with mcEDS-DSE was reported in 2013 (14), 14 patients have been described so far (15). Although the clinical picture of mcEDS-DSE was basically similar to that of mcEDS-CHST14, joint manifestations (recurrent/chronic joint dislocations, joint hypermobility), skin features (hyperextensibility, bruisability, fragility, atrophic scars), constipation, hypotonia, and motor developmental delay were significantly less common in mcEDS-DSE than mcEDS-CHST14 (15). Recently, the all Japan collaborative team reported an additional patient, in whose skin fibroblasts DS moieties on CS/DS proteoglycans were significantly decreased, but remained present. An ultrastructural study of the skin specimens, including cupromeronic blue-staining, revealed coexistence of normally assembled collagen fibrils with attached curved GAG chains and dispersed collagen fibrils with linear GAG chains from attached collagen fibrils across interfibrillar spaces to adjacent fibrils. A residual activity of DS-epi1, encoded by DSE, and/or compensation by DS-epi2, a minor homolog of DS-epi1, was suggested to contribute to the mild skin involvement through this "mosaic" pattern of collagen fibril assembly (15,16).
The author participated in the establishment of the diagnostic criteria (Table 1) (1) and the healthcare guideline (Table 2) (2,17) as a member of the EDS Society (https://www.ehlers-danlos.com) involving in the development and spread of medical care and research of EDS based on the international collaboration of patients and specialists. Novel clinical and basic evidences are expected be created and presented from the all Japan collaborative team including clinicians and researchers of various fields.
Table 1. Diagnostic criteria for mcEDS (1)
| Major criteria |
1. Congenital multiple contractures, characteristically adduction-flexion contractures and/or talipes equinovarus (clubfoot)
2. Characteristic craniofacial features, which are evident at birth or in early infancy
3. Characteristic cutaneous features including skin hyperextensibility, easy bruisability, skin fragility with atrophic scars, increased palmar wrinkling
|
Minor criteria |
1. Recurrent/chronic dislocations
2. Pectus deformities (flat, excavated)
3. Spinal deformities (scoliosis, kyphoscoliosis)
4. Peculiar fingers (tapering, slender, cylindrical)
5. Progressive talipes deformities (valgus, planus, cavum)
6. Large subcutaneous hematomas
7. Chronic constipation
8. Colonic diverticula
9. Pneumothorax/pneumohemothorax
10. Nephrolithiasis/cystolithiasis
11. Hydronephrosis
12. Cryptorchidism in males
13. Strabismus
14. Refractive errors (myopia, astigmatism)
15. Glaucoma/elevated intraocular pressure
|
Minimal criteria suggestive for mcEDS: |
At birth or in early childhood: Major criterion (1): Congenital multiple constractures, (2) characteristic craniofacial features
In adolescence and in adulthood: Major criterion (1): Congenital multiple constractures, (3) characteristic cutaneous features
Confirmatory molecular testing to detect bilallelic pathogenic variants in CHST14 or DSE is obligatory to reach a final diagnosis.
|
Table 2. Healthcare guidelines in mcEDS (2,17)
| Initial screening after diagnosis |
1. Evaluation of congenital cardiac, ocular, and renal abnormalities and hearing loss |
In infancy to childhood |
1. Orthopedic intervention (e.g., serial plaster casts or surgery) for talipes equinovarus
2. Physical therapy for motor developmental delay
3. Laxatives and/or enemas for constipation
4. Surgical fixation for cryptorchidism in males
5. Regular checkups for ophthalmological (strabismus, refractive errors, glaucoma), otological (otitis media with effusion, hearing impairment), urological (urination, bladder enlargement), and cardiovascular (valve abnormalities, aortic root dilation) problems
6. Prevention of skin lacerations, joint dislocations, or large subcutaneous hematomas associated with progressive foot deformities and trauma after walking independently
7. Intranasal administration of DDAVP (1-desamino-8-D-arginine vasopressin) or intravenous administration of factor VIII and von Willebrand factor (Confact F®︎) after trauma to prevent large subcutaneous hematomas
8. A wrist-type sphygmomanometer for hyperalgesia to pressure
|
From adolescence to adulthood |
1. Assessment of spinal deformities (scoliosis, kyphoscoliosis) and secondary sex characteristics (breast development in females and gonadal function in males) in adolescence
2. Treatment for emergency complications (e.g., pneumothorax or pneumohemothorax, large subcutaneous hematomas, and diverticular perforation)
3. Psychosocial support
|
Tomoki Kosho
(Department of Medical Genetics, Shinshu University School of Medicine; Center for Medical Genetics, Shinshu University Hospital; Division of Clinical Sequencing, Shinshu University School of Medicine; Research Center for Supports to Advanced Science, Shinshu University)
| References |
| (1) |
Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, Bloom L, Bowen JM, Brady AF, Burrows NP, Castori M, Cohen H, Colombi M, Demirdas S, De Backer J, De Paepe A, Fournel-Gigleux S, Frank M, Ghali N, Giunta C, Grahame R, Hakim A, Jeunemaitre X, Johnson D, Juul-Kristensen B, Kapferer-Seebacher I, Kazkaz H, Kosho T, Lavallee ME, Levy H, Mendoza-Londono R, Pepin M, Pope FM, Reinstein E, Robert L, Rohrbach M, Sanders L, Sobey GJ, Van Damme T, Vandersteen A, van Mourik C, Voermans N, Wheeldon N, Zschocke J, Tinkle B: The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 175, 8-26, 2017 |
| (2) |
Brady AF, Demirdas S, Fournel-Gigleux S, Ghali N, Giunta C, Kapferer-Seebacher I, Kosho T, Mendoza-Londono R, Pope MF, Rohrbach M, Van Damme T, Vandersteen A, van Mourik C, Voermans N, Zschocke J, Malfait F: The Ehlers-Danlos syndromes, rare types. Am. J. Med. Genet. C. Semin. Med. Genet. 175, 70-11, 2017 |
| (3) |
Malfait F, Castori M, Francomano CA, Giunta C, Kosho T, Byers PH: The Ehlers-Danlos syndromes. Nat. Rev. Dis. Primers 6, 64, 2020 |
| (4) |
Dündar M, Müller T, Zhang Q, Pan J, Steinmann B, Vodopiutz J, Gruber R, Sonoda T, Krabichler B, Utermann G, Baenziger JU, Zhang L, Janecke AR: Loss of dermatan-4-sulfotransferase 1 function results in adducted thumb-clubfoot syndrome. Am. J. Hum. Genet. 85, 873-882, 2009 |
| (5) |
Miyake N, Kosho T, Mizumoto S, Furuichi T, Hatamochi A, Nagashima Y, Arai E, Takahashi K, Kawamura R, Wakui K, Takahashi J, Kato H, Yasui H, Ishida T, Ohashi H, Nishimura G, Shiina M, Saitsu H, Tsurusaki Y, Doi H, Fukushima Y, Ikegawa S, Yamada S, Sugahara K, Matsumoto N: Loss-of-function mutations of CHST14 in a new type of Ehlers-Danlos syndrome. Hum. Mutat. 31, 966-974, 2010 |
| (6) |
Malfait F, Syx D, Vlummens P, Symoens S, Nampoothiri S, Hermanns-Lê T, Van Laer L, De Paepe A: Musculocontractural Ehlers-Danlos Syndrome (former EDS type VIB) and adducted thumb clubfoot syndrome (ATCS) represent a single clinical entity caused by mutations in the dermatan-4-sulfotransferase 1 encoding CHST14 gene. Hum. Mutat. 31, 1233-1239, 2010 |
| (7) |
Kosho T, Takahashi J, Ohashi H, Nishimura G, Kato H, Fukushima Y: Ehlers-Danlos syndrome type VIB with characteristic facies, decreased curvatures of the spinal column, and joint contractures in two unrelated girls. Am. J. Med. Genet. A 138A, 282-287, 2005 |
| (8) |
Kosho T, Miyake N, Hatamochi A, Takahashi J, Kato H, Miyahara T, Igawa Y, Yasui H, Ishida T, Ono K, Kosuda T, Inoue A, Kohyama M, Hattori T, Ohashi H, Nishimura G, Kawamura R, Wakui K, Fukushima Y, Matsumoto N: A new Ehlers-Danlos syndrome with craniofacial characteristics, multiple congenital contractures, progressive joint and skin laxity, and multisystem fragility-related manifestations. Am. J. Med. Genet. A 152A, 1333-1346, 2010 |
| (9) |
Shimizu K, Okamoto N, Miyake N, Taira K, Sato Y, Matsuda K, Akimaru N, Ohashi H, Wakui K, Fukushima Y, Matsumoto N, Kosho T: Delineation of dermatan 4-O-sulfotransferase 1 deficient Ehlers-Danlos syndrome: observation of two additional patients and comprehensive review of 20 reported patients. Am. J. Med. Genet. A 155A, 1949-1958, 2011 |
| (10) |
Kosho T, Miyake N, Mizumoto S, Hatamochi A, Fukushima Y, Yamada S, Sugahara K, Matsumoto N: A response to: loss of dermatan-4-sulfotransferase 1 (D4ST1/CHST14) function represents the first dermatan sulfate biosynthesis defect, "dermatan sulfate-deficient Adducted Thumb-Clubfoot Syndrome". Which name is appropriate, "Adducted Thumb-Clubfoot Syndrome" or "Ehlers-Danlos syndrome"? Hum. Mutat. 32, 1507-1509, 2011 |
| (11) |
Minatogawa M, Unzaki A, Morisaki H, Syx D, Sonoda T, Janecke AR, Slavotinek A, Voermans NC, Lacassie Y, Mendoza-Londono R, Wierenga KJ, Jayakar P, Gahl WA, Tifft CJ, Figuera LE, Hilhorst-Hofstee Y, Maugeri A, Ishikawa K, Kobayashi T, Aoki Y, Ohura T, Kawame H, Kono M, Mochida K, Tokorodani C, Kikkawa K, Morisaki T, Kobayashi T, Nakane T, Kubo A, Ranells JD, Migita O, Sobey G, Kaur A, Ishikawa M, Yamaguchi T, Matsumoto N, Malfait F, Miyake N, Kosho T: Clinical and molecular features of 66 patients with musculocontractural Ehlers-Danlos syndrome caused by pathogenic variants in CHST14 (mcEDS-CHST14). J. Med. Genet. 59, 865-877, 2022 |
| (12) |
Mizumoto S, Kosho T, Hatamochi A, Honda T, Yamaguchi T, Okamoto N, Miyake N, Yamada S, Sugahara K: Defect in dermatan sulfate in urine of patients with Ehlers-Danlos syndrome caused by a CHST14/D4ST1 deficiency. Clin. Biochem. 50, 670-677, 2017 |
| (13) |
Hirose T, Takahashi N, Tangkawattana P, Minaguchi J, Mizumoto S, Yamada S, Miyake N, Hayashi S, Hatamochi A, Nakayama J, Yamaguchi T, Hashimoto A, Nomura Y, Takehana K, Kosho T, Watanabe T: Structural alteration of glycosaminoglycan side chains and spatial disorganization of collagen networks in the skin of patients with mcEDS-CHST14. Biochim. Biophys. Acta. Gen. Subj. 1863, 623-631, 2019 |
| (14) |
Müller T, Mizumoto S, Suresh I, Komatsu Y, Vodopiutz J, Dundar M, Straub V, Lingenhel A, Melmer A, Lechner S, Zschocke J, Sugahara K, Janecke AR: Loss of dermatan sulfate epimerase (DSE) function results in musculocontractural Ehlers-Danlos syndrome. Hum. Mol. Genet. 22, 3761-3772, 2013 |
| (15) |
Minatogawa M, Hirose T, Mizumoto S, Yamaguchi T, Nagae C, Taki M, Yamada S, Watanabe T, Kosho T: Clinical and pathophysiological delineation of musculocontractural Ehlers-Danlos syndrome caused by dermatan sulfate epimerase deficiency (mcEDS-DSE): A detailed and comprehensive glycobiological and pathological investigation in a novel patient. Hum. Mutat. 43, 1829-1836, 2022 |
| (16) |
Syx D, Van Damme T, Symoens S, Maiburg MC, van de Laar I, Morton J, Suri M, Del Campo M, Hausser I, Hermanns-Lê T, De Paepe A, Malfait F: Genetic heterogeneity and clinical variability in musculocontractural Ehlers-Danlos syndrome caused by impaired dermatan sulfate biosynthesis. Hum. Mutat. 36, 535-547, 2015 |
| (17) |
Kosho T: CHST14/D4ST1 deficiency: New form of Ehlers-Danlos syndrome. Pediatr. Int. 8, 88-99, 2016 |
Oct. 12, 2023
|
|---|





