 この作品はクリエイティブ・コモンズ 表示 4.0 |
 |
|---|
糖鎖の分子動力学シミュレーション | |||||||||||
 |
分子の「形」は機能と密接に関係する。最近、人工知能プログラム「AlphaFold」がタンパク質の「形」を高い精度で予測することに成功して話題となった。分子の「形」がわかると次に知りたくなるのが「動き」である。 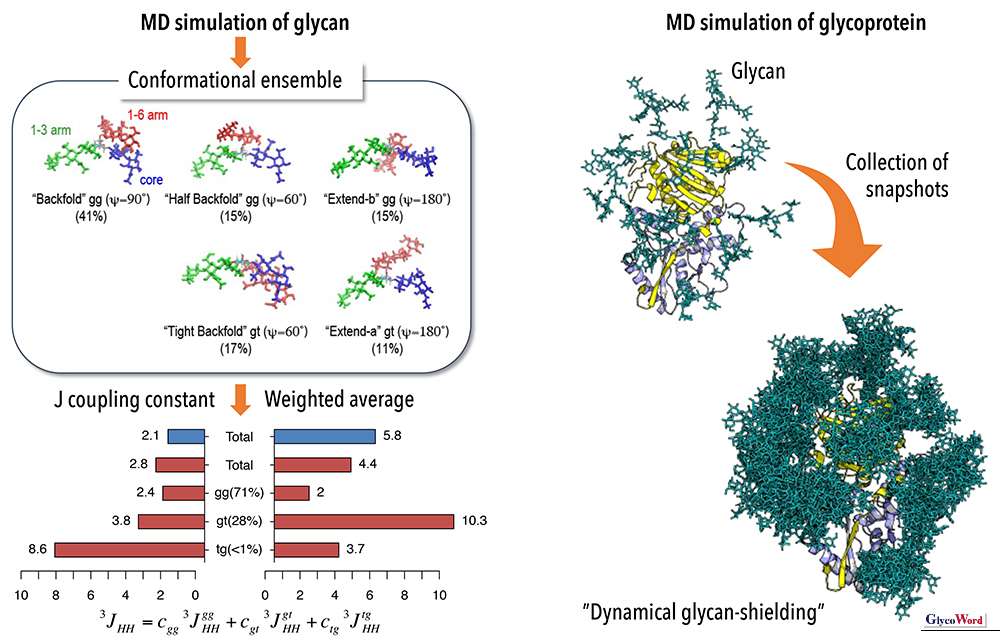
Fig. 1 李 秀栄(国立研究開発法人 医薬基盤・健康・栄養研究所)
2023年 6月15日   | ||||||||||


この作品はクリエイティブ・コモンズ 表示 4.0 |
|
|---|
糖鎖の分子動力学シミュレーション | |||||||||||
|
分子の「形」は機能と密接に関係する。最近、人工知能プログラム「AlphaFold」がタンパク質の「形」を高い精度で予測することに成功して話題となった。分子の「形」がわかると次に知りたくなるのが「動き」である。
Fig. 1 李 秀栄(国立研究開発法人 医薬基盤・健康・栄養研究所)
2023年 6月15日 | ||||||||||