 |
The "shape" of a molecule is closely related to its function. Recently, the artificial intelligence program "AlphaFold" succeeded in predicting the "shape" of proteins with high accuracy. Once we know the "shape" of a molecule, we next want to know its "motion".
Molecular dynamics simulation (hereafter MD simulation) is a physics-based method to probe the "motion" of molecules. It tracks changes in molecular structure (“shape”) over time by calculating interatomic forces and repeatedly solving Newton's equations of motion. Classical MD simulations, which are widely used for biomolecules, evaluate interatomic interactions at high-speed using empirical potential functions called force field. This allows enormous number of iterations for probing the biomolecular motion at atomic resolution with femtosecond timesteps. Several general-purpose program packages have been developed, including AMBER (https://ambermd.org), CHARMM (https://www.charmm.org), GROMACS (https://www.gromacs.org), and GENESIS (https://www.r-ccs.riken.jp/labs/cbrt/). Currently, these programs are widely used in academia and industry.
Glycans adopt various conformations due to the rotation of inter-residue glycosidic bonds. MD simulations can examine the details of dynamical structure, including fluctuations of molecules, rotation of glycosidic bonds, and accompanying changes in intra- and inter-molecular interactions, of whole molecules, which are difficult to see experimentally. However, since MD simulations track molecular motions in femtosecond timesteps, it is currently difficult to track millisecond or slower motions, such as transitions among rotamers. For example, gauche-trans transitions of α(1-6) glycosidic bonds are observed only a few times during 1 microsecond-long MD simulation (107 iterations). Several methods have been developed to search for various conformations by accelerating molecular motion. For example, the replica-exchange molecular dynamics (REMD) method runs multiple MD simulations in parallel at different temperatures, samples various conformations at high temperatures, and efficiently finds stable conformations at low temperatures by appropriate temperature exchange. Using such techniques, various rotamer conformations and their relative populations can be obtained with high statistical accuracy. By calculating the NOE (Nuclear Overhauser Effect) and/or spin coupling constants (J) across the glycosidic bond for each conformer, and taking their weighted average, the simulation results can be directly compared with NMR experiments to extract representative conformations. The concept of structural ensemble, which characterizes molecular motions with representative structures, makes it possible to explain the molecular mechanism of dynamical functions, such as glycan recognition by lectins, based on glycan conformation.
An initial structure and a force field are required for actual MD simulations. A force field is a set of energy functions and parameters representing interatomic interactions, and generally consists of bonded (bonds, angles, dihedral angles) and non-bonded (electrostatic and Lennard-Jones interactions) energy terms. The parameters in the functions (e.g. equilibrium internuclear distances and force constant values) are ordinary determined so as to reproduce the results of quantum chemical calculations and thermodynamic quantities. In addition to the general-purpose force fields for proteins, nucleic acids, and lipids, there exist several parameters that reproduce the structures unique to glycans (conformational diversity, puckering, anomeric effects, etc.). These involve CHARMM36, GLYCAM06 (AMBER), GROMOS 56A6CARBO, and OPLS-AA, and allow the simulations of not only free glycans, but also glycoconjugates, such as glycoproteins and glycolipids. The parameters commonly cover diverse carbohydrates with pyranose and furanose moieties, but the way to determine the parameters differs for each. The structures of glycans themselves can be predicted with similar accuracy, while those of glycoconjugates or even complex systems must be simulated with care. Hydration free energies may vary with the force field, and glycans can erroneously aggregate. A well-balanced description of mutual interactions between glycans-proteins/lipids-water molecules is important for such simulations. There have been ongoing efforts to improve the accuracy, for example by scaling non-bonding interactions for specific molecular pairs. In deciding which forcefield to use, one should pay attention to the latest developments, as well as the program and target simulation system of interest.
Modeling of the simulation system is particularly important for glycans which lack sufficient structure data. Recently, online tools such as GLYCAM-Web (https://glycam.org), CHARMM-GUI (https://www.charmm-gui.org/), GlyProt (http://www.glycosciences.de/modeling/) and scripts such as doGlycans (https://bitbucket.org/biophys-uh/doglycans/) are freely available. The initial model and input files for MD simulation are easily generated from the information of glycan types and sites to which they attach. Nowadays, the advances in the above-mentioned simulation environments make anyone possible to visualize glycans’ motion on a computer screen. By understanding the limitations due to model, force field accuracy, and simulation time (sampling), successful MD simulations in the future will reveal glycans’ dynamical structures underling various functions in detail.
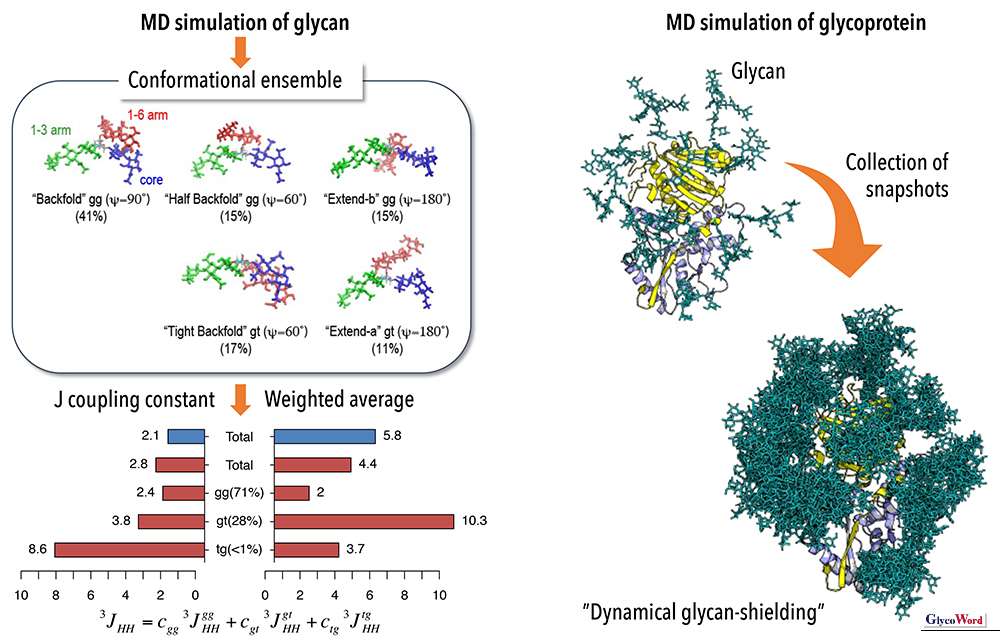
Fig. 1
Examples of MD simulations of glycans. Representative conformations of a N-glycan and J coupling constant (left) and the dynamical glycan-shield structure of a Lassa virus glycoprotein (right).
Suyong RE
(National Institutes of Biomedical Innovation, Health and Nutrition)
| References |
| (1) |
Re S, Yamaguchi Y, Sugita Y: Molecular Dynamics Simulation of Glycans. Trends Glycosci. Glycotechnol. 32, E113–18, 2020 |
| (2) |
Elisa F: Molecular Simulations of Complex Carbohydrates and Glycoconjugates. Curr. Opin. Chem. Biol. 69, 102175, 2022 |
| (3) |
Sugita Y, Kamiya M, Oshima H, Re S: Replica-Exchange Methods for Biomolecular Simulations. Methods Mol. Biol. 2022, 155–77, 2019 |
| (4) |
Sugita Y, Okamoto Y: Replica-Exchange Molecular Dynamics Method for Protein Folding. Chem. Phys. Lett. 314, 141-51, 1999 |
Jun. 15, 2023
|
|---|





