A. 発現タンパク質の設計
X線結晶構造解析の成功の可否はここで決まると言っても過言ではない。良質な結晶とは、結晶を構成する分子が如何に均一な立体構造を持ち、規則的に並んでいるかである。このため、タンパク質中の柔軟な領域を取り除き、運動性を極力低下させるように設計する必要がある。また、標的タンパク質が糖タンパク質であった場合、糖鎖は結晶化の妨げになるため、糖転移酵素をノックアウトした細胞を用いたり、糖加水分解酵素により糖鎖をトリミングする必要がある(2)。近年、深層学習を用いることにより、極めて高い精度を有するAlphaFold2等のタンパク質構造予測プログラムが開発された(3)。こうした予測プログラムの併用により、システマティックな発現タンパク質設計が可能になってきた。
B. 結晶化スクリーニング
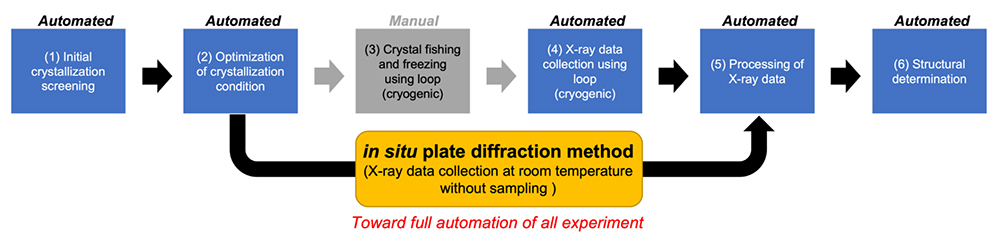
結晶化スクリーニングでは、500-1,000種程度の沈殿剤を用い微量分注機ロボットにより結晶化条件の探索を行うのが一般的である。近年の進展としては、結晶化スクリーニングと結晶の質の判定を同時に行う「in situプレート回折法」が挙げられる(4)。結晶の“見た目”と“質”は必ずしも相関しない。すなわち、実際に結晶にX線を当ててみるまで得たい情報は得られない。そのため、X線回折実験において、品質の不確かな膨大な数の結晶を結晶化プレートから取り出して回折実験に臨むことは稀では無い。一方、in situプレート回折法では、室温で結晶化プレートに対して直接X線を照射させることで、結晶の質を判定できるため、効率的かつ迅速な結晶化条件の最適化が可能になる。しかしながら、結晶位置の認識は現状では結晶学者による目によって判定しているため、現状は時間がかかり自動化に至っていない。今後、AI等を用いた結晶画像判定法の開発が課題である。
C. X線回折データ測定
近年、放射光ビームライン、X線検出器、解析ソフトウェア等の高度化や自動化が進み、測定や解析にかかる時間が大幅に短縮された。具体的には、100Kの極低温下で1〜20 μm角の微小なX線ビームを用いて結晶位置をスキャンし、最適条件下で全自動測定する。これにより、1つの結晶あたり5分程度で測定が完了する。また、複数の結晶からの回折データを自動的にクラスタリングし、良質なデータのみを選び出すソフトウェアが開発され、高難易度の膜タンパク質等の微小結晶からの構造解析が可能になっている(5)。
D. 初期位相決定および構造精密化
X線回折実験により得られる情報は、構造解析(具体的にはフーリエ合成)に必須な位相情報を失っているため、何かしらの方法により位相情報を決定する必要がある(1)。分子置換法は簡便な方法であり、最も広く用いられている。具体的には、まず本法では適当な類似構造(サーチモデル)を回転・並進させ、目的分子に対して重ね合わせたモデル座標を得る。次に、重ね合わせたモデル座標を元に位相を逆計算して、フーリエ合成により電子密度マップを得る。このとき、モデル座標が実構造に近ければ近いほど、より正確な位相情報すなわち明瞭な電子密度マップを与える。近年開発されたAlphaFold2(3)等のタンパク質構造予測プログラムは、高精度のモデル構造を作ることが出来るため、初期位相決定と構造精密化(実構造への修正作業)が大幅に加速している。
Blow D: Outline of Crystallography for Biologist. Oxford University Press, ISBN-10: 0198510519, 2002.
(2)
Kamiya Y, Satoh T, Kato K: Recent advances in glycoprotein production for structural biology: toward tailored design of glycoforms. Curr. Opin. Struct. Biol. 26, 44-53, 2014.
(3)
Jumper J, et al.: Highly accurate protein structure prediction with AlphaFold. Nature596, 583–589, 2021.
(4)
Martiel I, Olieric V, Caffrey M, Wang M: Practical approaches for in situ X-ray crystallography: from high-throughput screening to serial data collection. In K. Beis & G. Evans (Eds.), Chemical biology: Vol. 8. Protein crystallography: challenges and practical solutions (pp. 1-27). ISBN 978-1-78801-477-9, 2018
(5)
Yamashita K, Hirata K, Yamamoto M: KAMO: towards automated data processing for microcrystals. Acta Cryst. D74, 441-449, 2018.