 |
Outline for X-ray crystallography
Using X-ray crystallography, three-dimensional structures of a series of materials, including proteins, can be determined in ångström (0.1 nm)-level resolution based on the electron density (1). Scattering occurs upon X-ray irradiation to the materials, but the scattered light is usually very weak. Therefore, the accurate information is limited to the low-resolution data at about 10-100 Å. When the crystal is used, tons of molecules contribute to scattering (diffraction), and the crystal acts as a signal amplifier, thereby enabling high-resolution structure analysis. Theoretically, there are no upper or lower molecular weight limits in X-ray crystallography, and a wide variety of materials can be analyzed. However, highly flexible molecules exemplified by glycans are difficult to crystallize and are thus not subject to this method. In this review, structural analysis of proteins will be outlined.
Recent progress in X-ray crystallography
Flow of X-ray crystallography can be divided into [A] design of protein expression construct, [B] crystallization screening, [C] X-ray diffraction data collection, and [D] initial phase determination and structure refinement.
[A] Design of protein expression construct
It is not too much to say that the success or failure of X-ray crystallography is determined here. Good crystal can be defined by the uniform three-dimensional structure and regular arrangement of the molecules that make up the crystal. Therefore, it is necessary to design a protein in which the flexible regions are eliminated to reduce motility as much as possible. In addition, if the target protein is a glycoprotein, the flexible glycan prohibits the crystallization. Thus, it is needed to use cells in which glycosyltransferase(s) are knocked out or trim the glycan by glycosidase(s) (2). Recently, protein structure prediction programs such as AlphaFold2 with extremely high accuracy have been developed using deep learning (3). The combined use of such prediction programs has enabled systematic design of protein expression construct.
[B] Crystallization screening
In protein crystallization screening, searching for the crystallization conditions using a micro-dispenser robot with about 500-1,000 precipitants is common. Recent progress is the "in situ plate diffraction method" that simultaneously performs crystallization screening and crystal quality validation (4). In general, "appearance" and "quality" of crystal do not always have a positive correlation. In other words, the desired information cannot be obtained until the crystal is irradiated by X-ray. Therefore, we need to take out a huge number of crystals with uncertain quality from the crystallization plate and perform the X-ray diffraction experiment. In contrast, in situ plate diffraction method can validate the quality of crystal through direct irradiation of X-ray to the plate at room temperature, thereby enabling efficient and rapid optimization of crystallization conditions. However, recognition of crystal position in the plate is currently done by the crystallographer's eyes, which is time-consuming and thus has not yet been automated. The development of crystal image recognition methods using artificial intelligence will be an issue in the future.
[C] X-ray data collection
In recent years, the sophistication and automation of synchrotron radiation beamlines, X-ray detectors, and software have been greatly developed, shorting the time required for measurement and analysis. Specifically, crystal positions are scanned using a micro-focus X-ray beam with 1 to 20 μm square under a cryogenic temperature of about 100 K, and fully automated measurement can be performed under the optimized condition. This allows the measurement to be completed within about 5 minutes per crystal. In addition, the software has been developed that automatically clusters diffraction data from multiple crystals and selects only good quality data, thereby enabling structural analysis from very tiny crystals originating from highly challenging proteins such as membrane protein (5).
[D] Initial phase determination and structure refinement
Since information obtained by the X-ray diffraction experiment lacks phase information, which is essential for the structural analysis (i.e., Fourier synthesis), we need to determine that by certain methods (1). The molecular replacement method is the simplest and most widely used method. Specifically, model coordinates are obtained through superimposition with rotation and translation movements on the target molecule using an appropriate similar structure (called a “search model”). Next, the electron density map is obtained by Fourier synthesis using the back-calculated phase based on the superimposed model coordinates. A closer search model compared with the real structure will give more accurate phase information, i.e., a clearer electron density map. Recently developed protein structure prediction programs such as AlphaFold2 (3) can create very accurate model structure, which greatly accelerates initial phase determination and structure refinement, i.e., modification to the real structure.
Future perspective
X-ray crystallography is currently the most widely used method for structure determination of biological macromolecules owing to the short measurement and analysis times and no limitation of molecular weight. The only part of this method that has not been automated is the process of taking out crystals from the crystallization plate and freezing the crystals, which can be namely called craftsmanship. In the future, the entire experiments of X-ray crystallography will be fully automated through the advancement of room temperature measurement methods including in situ plate diffraction (Figure 1).
In recent years, there has been remarkable progress in cryo-EM analysis and protein structure prediction. The advantage of cryo-EM is that crystallization is not required. However, the disadvantage of this method is that it is very difficult to solve the structure of proteins with a molecular weight smaller than about 100 kDa, and the measurement time is long (approximately 150 times as compared with X-ray crystallography). In addition, protein structure prediction programs have the advantage of being able to accurately predict overall structure at the main-chain level of amino acid residues, but they are currently unable to predict glycoprotein structure with glycan modification and the binding mode of ligands such as sugar chains. Thus, since each method has its advantages and disadvantages, they would be used or combined according to sample characteristics and research purpose.
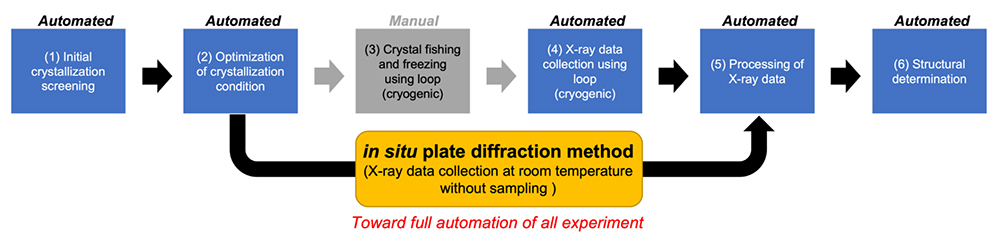
Figure 1.
Experimental flow and future perspective of X-ray crystallography
|
Tadashi Satoh
(AgroDesign Studios)
| References |
| (1) |
Blow D: Outline of Crystallography for Biologist. Oxford University Press, ISBN-10: 0198510519, 2002. |
| (2) |
Kamiya Y, Satoh T, Kato K: Recent advances in glycoprotein production for structural biology: toward tailored design of glycoforms. Curr. Opin. Struct. Biol. 26, 44-53, 2014. |
| (3) |
Jumper J, et al.: Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589, 2021. |
(4) |
Martiel I, Olieric V, Caffrey M, Wang M: Practical approaches for in situ X-ray crystallography: from high-throughput screening to serial data collection. In K. Beis & G. Evans (Eds.), Chemical biology: Vol. 8. Protein crystallography: challenges and practical solutions (pp. 1-27). ISBN 978-1-78801-477-9, 2018 |
(5) |
Yamashita K, Hirata K, Yamamoto M: KAMO: towards automated data processing for microcrystals. Acta Cryst. D74, 441-449, 2018. |
Jun. 15, 2023
|
|---|





